


A case of a long course of Osler–Weber–Rendu disease in a 65-year-old patient
- Government Institution L.T. Malaya Therapy National Institute of the National Academy of Medical Sciences of Ukraine, 61000, 2a Lyubov Malaya Ave., Kharkov, Ukraine
Abstract
The incidence of Osler–Weber–Rendu disease is low, ranging from 1 detected case per 50,000 to 1 per 100,000 population. The disease is hereditary, with autosomal dominant transmission, caused by pathogenic mutations in genes involved in angiogenesis. The disease has a pronounced clinical picture of multiple telangiectasias of the skin and mucous membranes and manifests as spontaneous bleeding. This scientific article presents a clinical analysis of a 65-year-old patient with a diagnosis of Osler–Weber–Rendu disease. Early identification of the manifestations of this disease and careful observation of the patient give a favorable prognosis of the course and prevent the development of severe complications.
Introduction
Osler–Weber–Rendu disease or hereditary hemorrhagic telangiectasia (HHT) is a rare systemic fibrovascular dysplasia. It is based on a disruption of the elastic and muscular layers of the vascular wall, which creates conditions for spontaneous ruptures and injuries1, 2. According to various sources, the prevalence of this disease ranges from 1 case per 50,000 to 1 per 100,000 population, and it has no gender characteristics3.
This pathology was first described in 1864 when H. G. Satton reported an illness that caused epistaxis in a patient with skin telangiectasias and internal bleeding4. In the following quarter of a century, several cases of HHT were described in the literature but could not be differentiated from hemophilia2, 5. In 1896, H. Rendu described the clinical case of a 52-year-old man with recurrent nosebleeds and telangiectasias on the face, body, lips, and soft palate, whose mother and brother also had episodes of nosebleeds5. Five years later, W. Osler reported three clinical cases of HHT. He drew attention to the hereditary characteristics of the disease; for the first time, he also described the injury of internal organs5, 6, 7. After another six years, F. Weber described the objective status of several patients with this disease; he noticed that they had vascular damage to the fingers, especially under the nails. Due to the contribution of these researchers, the condition is called Osler–Weber–Rendu disease or syndrome. The term HHT appeared only in 1909, formulated by A. Hans.
This disease is inherited in an autosomal dominant manner8, 9, 10 and caused by pathogenic mutations in genes involved in angiogenesis (endoglin, ACVRL1, SMAD4, GDF 2)7. However, around 20% of cases are due to sporadic mutations and have no family anamnesis8.
Currently, no effective HHT therapy exists11. The choice of treatment method is symptomatic and based on the degree of damage to the target organ5. Despite some success in developing experimental therapy to stabilize the abnormal vascular wall (using such drugs as tacrolimus and bevacizumab)9, 12, 13, HHT remains a potentially life-threatening pathology12.
Case presentation
We present the clinical case of Osler–Weber–Rendu disease in patient O., 65 years old, who was hospitalized in the Department of Gastroenterology and Therapy of the government institution L. T. Malaya Therapy National Institute of the National Academy of Medical Sciences of Ukraine on February 8, 2021. The white female patient had a height of 1.62 cm, weight of 70 kg, and BMI of 26.67 kg/m.
She complained of pain in the upper abdomen, nausea, vomiting of bile streaked with blood, heaviness after eating, nosebleeds, periodic increases in blood pressure up to 200/100 mmHg, numbness of the fingers and toes, cramps of the calf muscles, severe and extended pain in the left leg, headaches, dizziness, severe oral dryness and thirst mainly at night, severe general weakness, sleep disturbance, and pain in the upper part of the right ear auricle.
Medical history
According to oral information provided by the 65-year-old patient, she had experienced frequent nosebleeds since 2001, joined over the next few years by bleeding from the oral cavity. She was treated without examination, with the symptoms attributed to high blood pressure, which had also increased since 2001 to a maximum of 270/100 mmHg. She took antihypertensive drugs sporadically; during last year, she took lisinopril 5 mg once a day in the morning.
From 2002 to 2005, the patient was diagnosed with coronary artery disease, unstable (progressive) angina pectoris, postinfarction cardiosclerosis (2005), calcification of the left coronary artery, HF IIA with left ventricular diastolic dysfunction, FC ІІІ, hypertension grade III, left bundle branch block, type 2 diabetes mellitus of moderate severity (treated with insulin preparations since 2009), gastroesophageal reflux disease, chronic gastro-duodenitis (exacerbation phase), gallstone disease grade ІІІ, chronic calculous cholecystitis (remission phase), chronic pyelonephritis (remission phase), and microcalculi.
In 2007, she was hospitalized with a diagnosis of hemangiomatosis of the nasal mucosa, oral cavity, facial skin, tongue, and fingers, along with nosebleeds. Angiography of the external and internal carotid arteries was performed. Filling of hemangiomatous formations from the basin of the maxillary artery on the right and facial arteries on both sides was revealed. In addition, filling of hemangiomas of the nasal cavity from the basin of the ethmoidal internal carotid artery on the right was noted. Embolization was performed of the maxillary and facial arteries with emboli of 400–750 MCR.
In 2012, the transition of type 2 diabetes to a secondary insulin-dependent severe subcompensated form was diagnosed, with the development of universal diabetic angiopathy of the vessels of the lower extremities and diabetic nephropathy.
According to oral information provided by the patient, daily nosebleeds predominated, with blood loss from 100 to 400 mL. Blood loss of up to 250 mL was asymptomatic, but with larger volumes, general weakness and shortness of breath were observed. In most cases, the bleeding stopped spontaneously, but if this did not happen, the patient used hemostatic therapy herself of vikasol 10 mg once per day. The patient also noted increased bleeding and progressive deterioration during anterior nasal packing, which was performed twice.
Throughout her life, the patient periodically took enalapril, amlodipine, bisoprolol, cardiomagnyl, controloc, metformin, vitamin B6, folic acid, cardonate, and citrarginine.
During the last month, the patient’s general condition had significantly deteriorated, with nausea, vomiting of bile with blood streaks, and abdominal pain. She did not seek medical help and took omeprazole 20 mg in the morning herself. Due to her progressive deterioration, on February 5, 2021, the patient presented to the National Institute of Therapy of the L. T. Malaya National Academy of Medical Sciences of Ukraine. She underwent video esophagoduodenoscopy (showing distal esophagitis, congestive gastroduodenopathy, and multiple telangiectasias of the stomach; Osler–Weber–Randu disease) and ultrasound examination of the abdominal organs (showing diffuse changes in the liver, echo signs of chronic pancreatitis, cholelithiasis, and solitary microliths of the kidneys).
Family anamnesis
The patient had two pregnancies and two vaginal births, with profuse uterine bleeding after the second. According to oral information provided by the patient, her mother and mother’s sister experienced frequent nosebleeds. The patient’s brother was diagnosed with Osler–Weber–Rendu disease in 2008, which was genetically confirmed. The patient’s son and daughter experienced frequent nosebleeds. There was no significant allergic anamnesis. The patient denied smoking, alcohol use, or drug use.
Somatic status
The general condition was moderately severe. Consciousness was clear. The axillary body temperature was 36.6 °C. The skin and visible mucous membranes were pale; multiple telangiectasias were observed on the skin of the face, ears, lips, sternum, palms, and conjunctiva (Figure 1).

Extraoralvascular telangiectasia. A. Red-violet point and linear telangiectasias and papular telangiectasias on the red border of the lips and the skin of the face. B. On the tips of the fingers, multiple red-purple spotted lesions did not rise above the surface. C. Lesions on the conjunctiva of the eye. https://doi.org/10.6084/m9.figshare.17097008.v1

Intraoral manifestations of hereditary telangiectasia. A. Papular telangiectasias rising above the red border of the lips. B. Papular telangiectasias, protruding above the surface of the mucous membrane of the tongue. C. Papular telangiectasias on the ventral surface of the tongue. https://doi.org/10.6084/m9.figshare.17097011.v1
The patient noted tenderness of the skin surface of the left leg. The peripheral lymph nodes were not palpable. The thyroid gland was not enlarged. The musculoskeletal system was without visible abnormalities. Active and passive movements in the shoulder joints were painless and preserved in full volume. No peripheral edema was observed. On palpation, the chest was resistant and painless. A clear pulmonary sound was found by percussion over the entire surface of the lungs; vesicular breathing was observed by auscultation. The RR was 16 per minute. Examination of the atrial region revealed no pathological pulsation. By percussion, the borders of the heart were: right in the IV intercostal space along the right edge of the sternum, upper in the III intercostal space on the left, and left in the V intercostal space along the left midclavicular line. Heart sounds were rhythmic and muffled, with the accent of II tone above the aorta. The heart rate was 85 beats per minute, with satisfactory qualities. The ABP after 5 minutes at rest while sitting on the left hand was 180/100 mmHg. The abdomen was soft on palpation, sensitive in the right hypochondrium, epigastrium, and mesogastrium; the liver and spleen were not enlarged. The Pasternatsky symptom was negative on both sides. Urination was free and painless. One bowel movement occurred in 3 days, without pathological findings.
Consultation of the cardiologist. Diagnosis: Ischemic heart disease. Stable exertional angina FC II. Postinfarction (2005) cardiosclerosis. Hypertensive disease stage III, grade 3, HF stage IIA with diastolic dysfunction of the left ventricle, FC III, left bundle branch block.
Consultation of the endocrinologist. Diagnosis: Diabetes mellitus type 2, secondarily insulin-dependent in the stage of decompensation, severe form. Diffuse nodular goiter. Euthyroid state.
Consultation of the neurologist. Diagnosis: Dyscirculatory encephalopathy of mixed genesis, grade II.
Consultation of the ophthalmologist. Diagnosis: Hypermetropia, incipient cataract in both eyes. Neuroproliferative diabetic retinopathy in both eyes. Hypertonic retinal angiopathy.
Consultation of the otolaryngologist. Diagnosis: Perichondritis of the right ear auricle. Recurrent nosebleeds.
Consultation of the dentist. The red border of the lips was dry and pale, with pronounced multiple punctate telangiectasias of red-violet color. In the area of the lower lip, brightly colored round nodules with a diameter of around 5 mm were found, protruding above the surface and painless on palpation (Figure 2А). The mucous membrane of the oral cavity was pale pink, swollen with dental prints and with a pronounced vascular pattern. The tongue was enlarged and bright pink, with mobility preserved. On the dorsal and lateral surfaces were red-violet papular formations, with a diameter of 5 to 7 mm, protruding above the surface (Figure 2В,C). Palpation of lesions was painless. The gums were hyperemic, edematous, and loose, with bleeding on probing and periodontal pockets with exudative contents. Abundant soft and dense dental plaque and calculus were found. Oral hygiene was unsatisfactory. On examination, saliva was insufficient, frothy, and located mainly in the retromolar region. The salivation rate was reduced (0.1 mL per minute).

Localization of vascular telangiectasias on the mucous membrane of the tongue. A. The gums are pale pink and dense. B. The tongue is moist, with less plaque. https://doi.org/10.6084/m9.figshare.17097014.v1
Additional examinations
Clinical blood test (08.02.2021): Hb 104 g/L, erythrocytes 4.32*10/L, increased ESR 40 mm/h; biochemical blood test (08.02.2021): increased glucose up to 18.53 mmol/L. Triiodothyronine 3 free 4.4 pmol/L, free T4 17 pmol/L, TSH 1.92 mIU/L (08.02.2021). Glycosylated hemoglobin 11.02% (10.02.2021); АРО-А1 1.2 g/L (10.02.2021); C-peptide 3.45 ng/mL (10.02.2021).
Clinical analysis of urine (08.02.2021): amount 70.0, light yellow color. Transparent, pH 7.0, specific gravity 1010, protein 0.010, glucose 1%, leukocytes 1–4 in the field of view, flat single epithelium, a little mucus.
Coprogram. The feces are formed, soft, and brown. The occult blood reaction was negative. Unchanged muscle fibers were absent. Changed muscle fibers were rare. Little vegetable fiber was undigested. Rare iodophilic flora, protozoan helminth eggs, and fungal elements were not found.
Electrocardiogram. The 12-lead supine position sinus tachycardia heart rate was 91 per minute. Complete left bundle branch block. Atrial block I degree. Cicatricial changes in the anterior-septal-apical-lateral wall of the left ventricle.
Chest X-ray. Signs of left ventricular hypertrophy and aortic induration. Calcification of the left coronary artery and aortic arch.
Transthoracic ultrasound examination of the heart. Fibrosis of the cusps of the aortic and mitral valves. Accessory chord of the left ventricle. No areas of dyssynergia were found. The contractile function of the heart (EF 59%) was good. Diastolic dysfunction of restrictive type.
Ultrasound examination of the thyroid gland. Nodular goiter stage I.
Principal diagnosis
Osler–Weber–Rendu disease: multiple telangiectasias of the body of the stomach; recurrent nosebleeds; telangiectasias of the mucous membranes of the oral cavity, lips, ears, facial skin, and palms; hereditary history (mother and brother; according to the criteria of Curacao).
Secondary diagnoses
Gastroesophageal reflux disease with distal esophagitis. Cholelithiasis. Chronic calculous cholecystitis. Chronic secondary pancreatitis, with pain and dyspeptic syndrome in the acute stage, disrupting the external and intrasecretory function of the pancreas.
Diabetes mellitus type 2, secondary insulin-dependent in the stage of decompensation, severe form. Diffuse nodular goiter. Euthyroidism.
Ischemic heart disease. Stable exertional angina FC II. Postinfarction (2005) cardiosclerosis. Hypertension stage III, grade 3. SN II stage A with preserved LVEF. Dyscirculatory encephalopathy of mixed genesis, grade II.
Hyperopia, incipient cataract in both eyes. Neuroproliferative diabetic retinopathy in both eyes. Hypertensive retinal angiopathy. Perichondritis of the right ear auricle, xerostomia, and moderate chronic generalized periodontitis.
Medical treatment
-
Omeprazole 40 mg + saline 1,000 mL intravenous drip, ursofalk 250 mg in the evening.
-
Corvasan 12.5 mg twice a day in the morning and evening, depending on blood pressure and heart rate; Bi-Prenelia 4/5 mg in the evening, atorvastatin 20 mg before bedtime.
-
Tardyferon 80 mg twice a day in the morning and evening, depending on clinical blood tests.
-
Insulin therapy: 12 Mixtard before breakfast, 10 Mixtard before dinner, Diaformin 1,000 mg after dinner, Dialipon 1,200 mg intravenously + saline 2,000 mL, vitacertin 2 mL intramuscularly, ascorutin 1 tablet twice a day in the morning and evening, for 1 month.
-
Visiloton 1 drop three times a day in both eyes, then taufon 4% 1 drop three times a day in both eyes.
-
Compress on the ear auricle: levomekol + dimexide, aqua-nasal spray with D-panthenol and hyaluronic acid in the nose 4–6 times a day.
-
Professional oral hygiene with training in teeth cleaning.
-
The day before invasive dental treatment, amoxicillin 500 mg was recommended to prevent the development of a brain abscess. Blood pressure was measured three times per visit. Treatment was carried out with an upright chair position to prevent nose and pulmonary bleeding, hypoxia, and cerebral embolism.
At the next visit, the patient noted an improvement in the condition in the oral cavity, with decreased dryness and a feeling of cleanliness (Figure 3A,B). We were unpleasantly surprised to find new lesions during stomatoscopy that were not detected during the examination, as well as new lesions after the application of OptraGate in the course of treatment (Figure 4A,B).
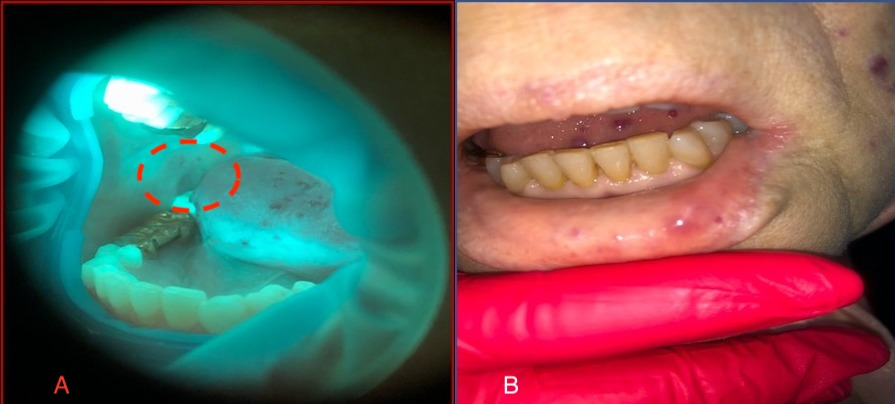
Pathological changes in the oral mucosa during the patient's sanitation. A.New lesions in the region of the retromolar space. B. New lesion on the lip mucosa after use of OptraGate. https://doi.org/10.6084/m9.figshare.17097020.v1
Discussion
This clinical case illustrates the non-alertness of doctors when examining this patient. From a vivid manifestation, despite the characteristic clinical picture and presence of family history, the diagnosis was established only after 7 years. Doctors could have been misled by the late onset of the disease at age 65 because, as a rule, the clinical debut of HHT is in adolescence. However, according to modern understanding, it can manifest at any age14.
The diagnosis of HHT is now based on the Curacao criteria established by the Scientific Department of the International Foundation for Hereditary Hemorrhagic Telangiectasia in 19999, 15, 16, 17: spontaneous and recurrent epistaxis, multiple telangiectasias in characteristic sites (lips, oropharynx, fingers, and nose), injury of internal organs (gastrointestinal telangiectasia with or without bleeding; pulmonary, hepatic, head, and spinal arteriovenous malformations), and family history (first-degree relative). The presence of three of these criteria defines the diagnosis of HHT as “reliable,” two criteria as “probable,” and one criterion as “doubtful”15. Therefore, despite the late onset of the disease, the attention of doctors should have been drawn to the characteristic episodes of nosebleeds, bleeding from the oral cavity, and postpartum uterine bleeding in combination with the family history of a first-degree relative. According to the Curacao criteria, these made the diagnosis of HHT already probable at the time of disease manifestation.
The disease directly impacts life expectancy; the average is lower in comparison with the general population18. With no family history (parents did not have HHT), the peak mortality of patients occurs at the age of 70 – 79 years, whereas in patients with heredity, the average life expectancy is 63.2 years14. Therefore, attention is drawn to the patient’s older age (65 years), despite the severe course of the disease (profuse daily nosebleeds, bleeding from the oral cavity, and the presence of a family history) and serious comorbidities. In this context, in our opinion, the patient’s having successfully vaginally delivered two children is a happy coincidence, given the concomitant presence of very high blood pressure. This again emphasizes the importance of timely diagnosis of Osler–Weber–Rendu disease in the absence of routine vigilance of the doctor, due to the low prevalence of the disease. The description of this case aims to improve the understanding of this pathology by general practitioners and, accordingly, increase its timely detection.
Conclusion
The relatively rare occurrence of the disease and insufficient awareness of doctors leads to frequent diagnostic errors. In addition, the symptoms and first complaints of patients with HHT vary. To improve the diagnosis of this rare and potentially serious disease, a careful approach is required to identify its characteristic symptoms, and knowledge of the dermatological aspects is important.
The prognosis is favorable in cases where bleeding can be controlled, and the reported mortality from complications is less than 10% of cases. Treatment for this disorder is only supportive and aims to relieve symptoms, treat complications, and prevent disease progression. Although in most cases, except recurrent nosebleeds, symptoms are absent or accompanied by minor complications, conducting an extensive examination is important to determine the location of the arteriovenous malformation and the systemic consequences of this disease. Therefore, early recognition of arteriovenous malformation of the liver, brain, and lungs and careful monitoring are important to prevent the development of severe complications and connected with a more favorable prognosis.Dental treatment can only be symptomatic and aimed at eliminating foci of chroniosepsis, which will significantly improve the quality of life of these patients.
Abbreviations
None.
Acknowledgments
None.
Author’s contributions
All authors have sufficiently contributed to the study, and agreed with the results and conclusions. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki. The institutional review board approved the study, and all participants provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.