

Long QT syndrome: Identification of a novel de novo mutation of calmodulin in a newborn girl
- Department of Biology, Dalat University, Da Lat City, Viet Nam
- Functional Genomics Unit, DNA Medical Technology Company, Ho Chi Minh City, Viet Nam
- Center for Molecular Biomedicine, University of Medicine and Pharmacy, Ho Chi Minh City, Viet Nam
- Department of Molecular Genetics, City Children’s Hospital, Ho Chi Minh City, Viet Nam
- Radiation Technology Center, Nuclear Research Institute, Da Lat City, Viet Nam
Abstract
Background: Long QT syndrome (LQTS) is a genetically and phenotypically heterogeneous disorder of ventricular myocardial repolarization that is typically characterized by prolongation of the heart rate-corrected QT interval on a 12-lead electrocardiogram (ECG). LQTS has a complicated etiology since great genotypic and phenotypic overlapping among LQTSs limits the establishment of a diagnosis based solely on clinical features.
Case presentation: Here, we describe a Vietnamese newborn with a novel calmodulin (CALM2) variant. The patient was referred to the Department of Cardiology at Children's Hospital 2 in Ho Chi Minh City, Vietnam for multiple clinical cardiac presentations. The patient's family history was negative for cardiovascular diseases; however, she suffered from recurrent syncope and QT interval prolongation on resting in a 12-lead surface ECG. A fetal echocardiogram at 29 weeks detected signs of congenital heart disease. The newborn patient then suffered from sudden cardiac arrest and required cardiopulmonary resuscitation. Two weeks later, the patient suffered from torsade de pointes for the third time and required electrical shocks. An endotracheal tube was placed through the mouth into the trachea to facilitate breathing. Unfortunately, the patient expired at 3 months of age due to serious inflammatory issues. Whole exome sequencing (WES) — a technique used to determine variations in all coding regions (exons) of the known genes — validated a total of four rare variants in four cardiomyopathy (CM) genes, the CALM2, DSP, MYBPC3, and SPEG genes. We identified a novel de novo heterozygous missense mutation, c.280G > T (p.Asp94Tyr), in the CALM2 gene (NM_001305626) encoding calmodulin, which is a ubiquitous calcium-binding protein that is important for a myriad of intracellular signaling events of cardiac function.
Conclusion: The results obtained in this study support the ``pan-cardiomyopathy panel'' approach, in which the molecular diagnosis of LQTS, the early identification of arrhythmia development, and the improved clinical management of cardiovascular disease patients are applied.
Trial registration: The procedures were reviewed and approved by the Ethical Committee of the University of Medicine and Pharmacy, Ho Chi Minh City, Vietnam, under the number UMP-VN 2018-10a12.
Introduction
Long QT syndrome (LQTS) is a genetically and phenotypically heterogeneous disorder of ventricular myocardial repolarization that is typically characterized by prolongation of the heart rate-corrected QT interval (QTc) on a 12-lead electrocardiogram (ECG)1, 2. This syndrome leads to torsadogenic syncope, seizures, aborted cardiac arrest, and sudden cardiac death (SCD)3, 4. LQTS is associated with autopsy-negative sudden death in the young, sudden infant death syndrome (SIDS), and even stillbirths5, 6, 7. Recently, mutations have been identified in at least 17 LQTS-susceptibility genes8, 9. LQTS has a complicated etiology, including multiple non-genetic and genetic causes10. Moreover, LQTS includes the complexity associated with genotype-phenotype correlation because not all mutations on the same gene produce a similar clinical phenotype11. The impact on clinical manifestations is unpredictable, as neither the localization of a mutation nor its cellular electrophysiological effect is associated with the phenotypes12. In certain cases, different LQTS subtypes have the same cardiac phenotype, and an identical mutation can cause symptomatic and asymptomatic LQTSs and other syndromes, leading to difficulties regarding the management of patients and a requirement for multiple ECG recordings11, 13, 14. Consequently, great genotypic and phenotypic overlapping among LQTSs limits the establishment of a diagnosis based solely on clinical features. Therefore, a specific diagnosis and prognosis for LQTS patients are reliant on next-generation sequencing (NGS) technology and genome-wide association studies.
NGS refers to high-throughput DNA sequencing technology that generates data on multiple DNA fragments in a single reaction15. NGS methods, including whole-exome sequencing (WES) and whole-genome sequencing (WGS), have rapidly become scalable tools that generate a molecular diagnosis for Mendelian genetic disorders15, 16, 17. The exome, which is composed of exons, comprises approximately 1% of the total genome and harbors 85% of interpretable mutations that result in clinical phenotypes18, 19. Accordingly, the identification and sequencing of this 1% coding region, which has less repetitive sequences than the noncoding regions, is computationally and analytically less intense15, 16. WES overcomes the limitations of Sanger sequencing methods because of its speed, cost, and accuracy15, 16, 17, 18, 19. When used as a first-line test, the diagnostic yield of WES is cost-effective and has become a powerful tool in identifying the causal variant(s) for a disease of interest17.
Since cardiovascular diseases (CVDs) are highly heritable, human genetic variation is considered to contribute to the detection of individuals who are at risk of developing cardiomyopathy (CM), heart failure, arrhythmias, and vascular diseases.
This present study reports on the family of a newborn girl with multiple clinical cardiac presentations who was diagnosed with LQTS type 3 (as she carried a heterozygous nonsynonymous mutation, c.280G > T (p.Asp94Tyr), in the calmodulin-2 () gene. The results of this study support the genetic etiology for LQTS and highlight the need for screening of the gene in congenital cardiovascular probands.
Case presentation
Materials and methods
Patient data
The present study reports on the Vietnamese family of a newborn index patient who was diagnosed with CVD by specialist doctors at Children’s Hospital 2 in Ho Chi Minh City, Viet Nam. The QT intervals were measured on the ECG in lead II or V5, and the QTc was corrected for using Bazett’s formula. The LQTS diagnosis was based on the symptoms and QTc.
The clinical manifestation, laboratory results (including ECG) and echocardiography, application of drugs, and prognosis of the patient were reviewed. The parents of the patient provided consent for genetic analysis. The study was approved by the local ethics committee and complied with the standards set by the hospital.
Library preparation and sequencing
Genomic DNA was isolated from white blood cells using standard techniques (DNA Extraction Kit, Qiagen, Hilden, Germany). The DNA fragments were ligated with adaptors and two paired-end DNA libraries with an insert size of 100 – 900 bp. After enrichment by polymerase chain reaction (PCR), the DNA libraries were prepared for cardiac gene enrichment re-sequencing on a HiSeq 4000 next-generation sequencing system according to the manufacturer’s instructions (TruSeq 3000/4000 SBS Kit v3 — protocol HiSeq 3000/4000 system user guide, part # 15066496, Rev. A HSC 3.3.20). The patient was screened for variants in 142 CM-associated genes using the TruSeq™ Cardiomyopathy gene panel, which resulted in 150-bp paired-end sequencing reads and at least a 100-fold average sequencing depth for each sample.
Alignment and mapping
The low-quality reads were discarded from raw data to generate clean reads. The clean reads were then aligned to the human reference genome UCSC Genome Build hg19. The Illumina automated workflow was used for the alignment of the reads and base calling: the Burrows-Wheeler Aligner (BWA), Genome Analysis Toolkit (GATK), and Sequence Alignment Map (SAMtools).
Variant annotation and filtering
For the annotation of variants, we used the ANNOVAR and VEP software programs. The variants were then computationally compared with a list of reported pathogenic variations from the Human Gene Mutation Database (HGMD, professional version). The extremely rare variants located in the exon and consensus splice sequences (± 1 and 2 positions) were filtered using a global minor allele frequency (MAF) of ≤ 0.0005. The variants that were not in the HGMD and synonymous and intronic variants that were greater than 15 bp from the exon boundaries were not considered.
Variant classification
The guidelines of the American College of Medical Genetics and Genomics (ACMG) were used for variant classification to assess dominant single-gene mutations with clear clinical impact on CMs (cardiomyopathies). The Exome Aggregation Consortium (ExAC) browser was used as a reference for the allele frequencies. The missense variant was assessed using a variety of prediction tools, including deleterious annotation of genetic variants using neural networks (DANN), Genomic Evolutionary Rate Profiling (GERP), MutationTaster, Protein Variation Effect Analyzer (PROVEAN), and Sorting Intolerant from Tolerant (SIFT). The degree of conservation of the affected residue was measured by multiple ortholog alignment using Alamut software (Interactive Biosoftware, Rouen, France).
Sanger sequencing
All variants of interest were confirmed using Sanger sequencing. The primers for the identified variants were designed using Primer3. The PCR primer pair used to amplify the products were CALM2-F: 5’-CGCCATGTGATGACAAACCT-3’ and CALM2-R: 5’-GTTCCCTAACAAAGAGCCTC-3’. Direct sequencing of the PCR products was performed in an ABI3130XL sequencer (Applied Biosystems) according to the manufacturer’s protocol.
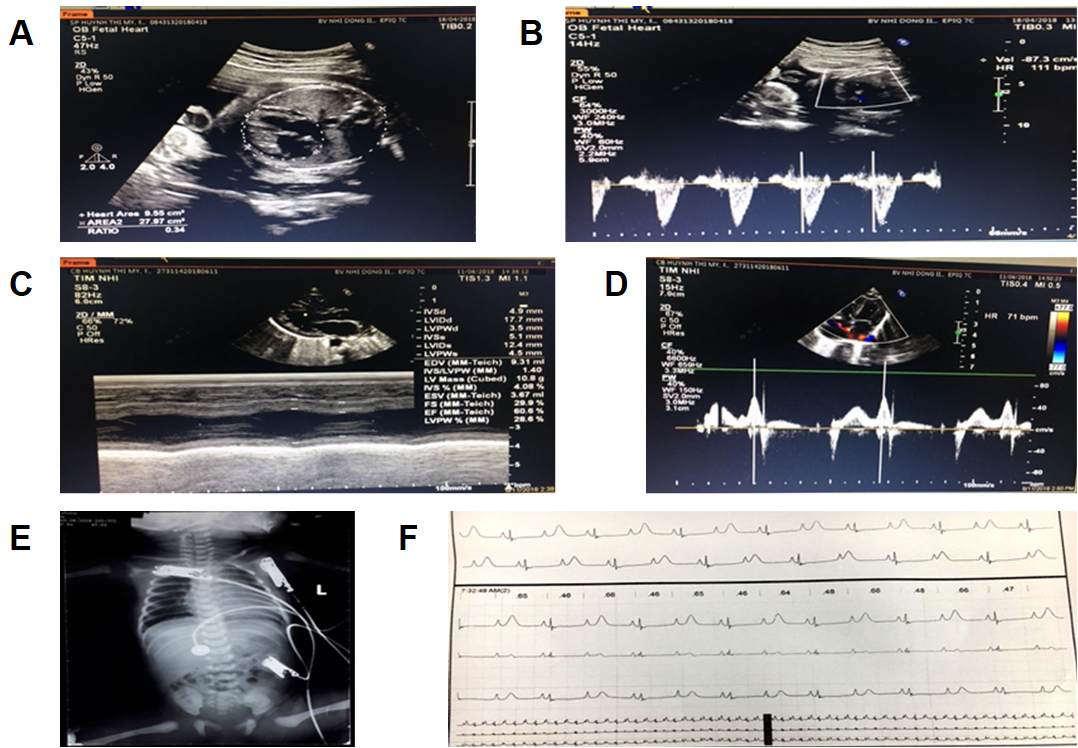
Patient clinical information. A & B) Fetal echocardiographies at 29 weeks with congenital heart disease signs during the prenatal period. C & D) Echocardiographies of the patient’s first day of birth showed depressed myocardiac function with EF 60.6% and ventricular abnormalities. E) Digital X-ray image of anteroposterior view of chest showing cardiac morphology with cardiomegaly. F) Electrocardiography showed a markedly prolonged QTc interval of 720 ms. https://doi.org/10.6084/m9.figshare.18865457.v1
Results
Patient clinical information
The subject of this study was a newborn baby girl who suffered from recurrent syncope and QT interval prolongation on a resting 12-lead surface ECG. The patient, who was born to healthy parents, was the family's only child. Her family history was negative for SCD, and both parents were asymptomatic regarding CVD and showed normal ECGs (data not shown).
A fetal echocardiogram at 29 weeks detected signs of congenital heart disease (CHD), including sinus arrhythmia with slowing of the heart rate, macrocardia, and highly inconsistent echocardiographic grading of aortic stenosis (AS) which was represented by bicuspid aortic valve (BAV) disease (Figure 1 A and B).
On June 10, 2018, the primigravida mother had a premature rupture of membranes (PROM) at 36 weeks of gestation. After amniorrhexis, a baby girl weighing 2,300 grams, who was in a breech presentation, was delivered in good condition by emergency caesarean section.
The neonate's diagnosis at the time of hospital admission showed a conscious state; however, her sinus rhythm was slow at a rate of 60 beats per minute (bpm).
During hospitalization, the low-birth-weight infant was well breastfed and reached a weight of 2,450 grams after one week. However, the electrocardiography showed a markedly prolonged QTc interval of 700 ms. Sinus arrhythmia was also included in the clinical record as the heart rate ranged from 60 to 100 bpm. Moreover, an inconsistent T wave alternans (TWA) signal was detected using the standard clinical leads. Echocardiograms showed a 2:1 atrioventricular (AV) block, left ventricular non-compaction cardiomyopathy (LVNC) symptoms, and a cardiac ejection fraction (EF) that ranged from 54 to 60.6% (Figure 1C and D). Based on these clinical symptoms, the cardiologists strongly suspected LQTS type 3. The patient was then subdermally implanted with a single-chamber pacemaker beneath the collarbone which was connected to the heart by one wire. A beta-blocker and mexiletine were administered to the patient daily.
On June 29, 2018, the patient suffered from sudden cardiac arrest (SCA) and required cardiopulmonary resuscitation (CPR) for 15 minutes to provide oxygenation and circulation to the body. After this intensive medical care, propranodol was administered to the patient. A low heart rate of 50 to 60 bpm was identified. During the subsequent days, the patient required a single-chamber pacemaker again and was continually medicated with propranodol. The heart rate increased to approximately 70 bpm.
Twelve days later, on July 10, 2018, the patient presented to the emergency department due to choking (on milk), cyanosis, and cardiac arrest, and required electrical shock (ES) and respiratory devices. The heart ceased to beat due to torsade de pointes (TdP), which occurred twice and lasted for 3 seconds each time. An ECG Holter monitor showed the occurrence of polymorphic ventricular tachycardia and the heart rate ranged from 80 to 115 bpm. A digital X-ray image showed cardiac enlargement (Figure 1E). Moreover, the patient's QTc had increased to 720 ms (Figure 1F). Thus, the patient was medicated with adrenaline and magnesium sulfate. Two weeks later, the patient suffered from TdP for the third time and required ES and endotracheal intubation (EI). An endotracheal tube was placed through the mouth into the trachea to facilitate breathing. Unfortunately, the patient expired at 3 months of age on September 10, 2018, due to serious inflammatory issues.
Pre-allignment statistics
|
Total number of reads |
27,514,276 |
|---|---|
|
Average read length (bp) |
151.0 |
|
Total yield (Mbp) |
4154 |
|
SNP |
88,187 |
|
Indel |
182 |
|
GC content (%) |
49 |
Statistic variants
|
No |
Gene |
Chromosome |
Exonic Function |
HGVS.c |
HGVS.p |
|---|---|---|---|---|---|
|
1 |
CALM2 |
2 |
Nonsynonymous SNV |
c.G280T |
p.D94Y |
|
2 |
DSP |
6 |
Nonsynonymous SNV |
c.A3646G |
p.I1216V |
|
3 |
MYBPC3 |
11 |
Nonsynonymous SNV |
c.C529T |
p.R177C |
|
4 |
SPEG |
2 |
Nonsynonymous SNV |
c.A8710G |
p.T2904A |
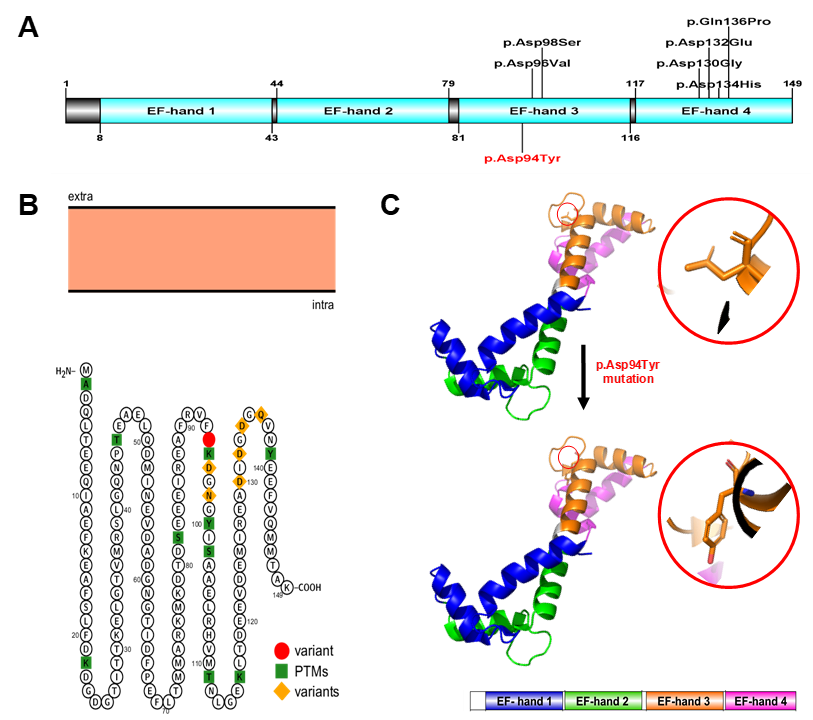
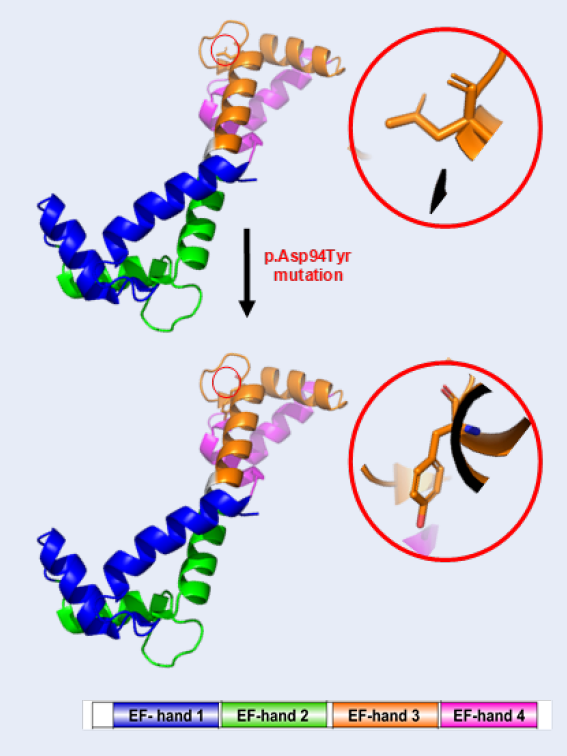
The novel CALM2 mutation. A) Schematic representation of domain structure of calmodulin protein (CaM). CaM, which is encoded by CALM2 gene, is a 21,689-dalton protein of 149 amino acids. CaM is an EF-hand protein composed of two calcium-binding EF-hand motifs (black) in its N-domain and two in its C-domain with a calcium-binding site on each domain (reported by Prosite). Positions of known pathogenic variants over the length of the canonical CaM are shown in black and our novel mutation is shown in red. B) General model of human CaM highlighting its most relevant features. CaM is an intracellular target of the secondary messenger calcium ion. The binding of calcium is required for the activation of CaM. Our novel mutation site is shown in red circle. Several previously known variants are shown in yellow. PTMs: post-translational modifications. The cell membrane is in light orange. The graph was generated using Protter v1.0. C) 3D-structures of CaM model. The positions of codon 94 are circled in red. The structures were created using PyMOL (http://www.pymol.org). https://doi.org/10.6084/m9.figshare.18865466.v1
Identification of a novel heterozygous -gene mutation
Genetic testing could help to increase the diagnostic accuracy and facilitate the clinical assessment and appropriate therapy of patients who have heart failure or arrhythmias, thus, preventing SCD. Therefore, in the present study, WES was performed on the patient to identify genetic variants.
The genetic data obtained for the patient are shown in
To further analyze the mutations and identify potential genes, the potential candidates were condensed and included 142 genes that have been linked to CM. These genes were further filtered based on the mapping quality MQ ≥ 40, prediction of protein alteration, missense variants, and absence from the SNP database of ADS. As a result, four variants in different genes were identified (
The results demonstrated that the WES identified a novel heterozygous missense variant in the gene (NM_001305626). The variant c.280G > T (p.Asp94Tyr) belonged to the exon 4, where a single nucleotide substitution occurred: G was replaced by T at position 280 in the cDNA, leading to the replacement of aspartic acid (Asp, D) by tyrosine (Tyr, Y) at the position 94 on the calmodulin protein (CaM) (Figure 2). (p.Asp94Tyr) produces the substitution of an acidic amino acid with a polar non-charged one. The physicochemical properties of Asp and Tyr are very different.
In this case, the only potentially pathogenic variant observed in the patient was c.280G > T (p.Asp94Tyr) in (
The human CaM is a small 149-amino acid protein that comprises two Ca-binding lobes at its N- and C-terminus, each containing two EF-hand structural motifs with Ca-binding properties. The novel c.280G > T (p.Asp94Tyr) mutation is located at the central EF-hand calcium-binding 3 domain (Figure 2A). Importantly, the C-terminals EF-hands 3 and 4 possess a higher Ca-binding affinity than those in the N-terminus and cluster various calmodulinopathy-causing mutations that express multiple arrhythmic phenotypes in the young, suggesting the functional importance of specific topological domains that appear to be intolerant to genetic variation20.
|
Detected variant |
Coding impact |
ACMG classification |
|
|---|---|---|---|
|
CALM2:NM_001305626:exon4:c.G280T:p.D94Y |
Nonsynonymous SNV |
Likely Pathogenic (PM1, PM2, PM5, PP3) |
DANN (0.9899) GERP (5.7399) MutationTaster (D) PROVEAN (D) SIFT (D) |
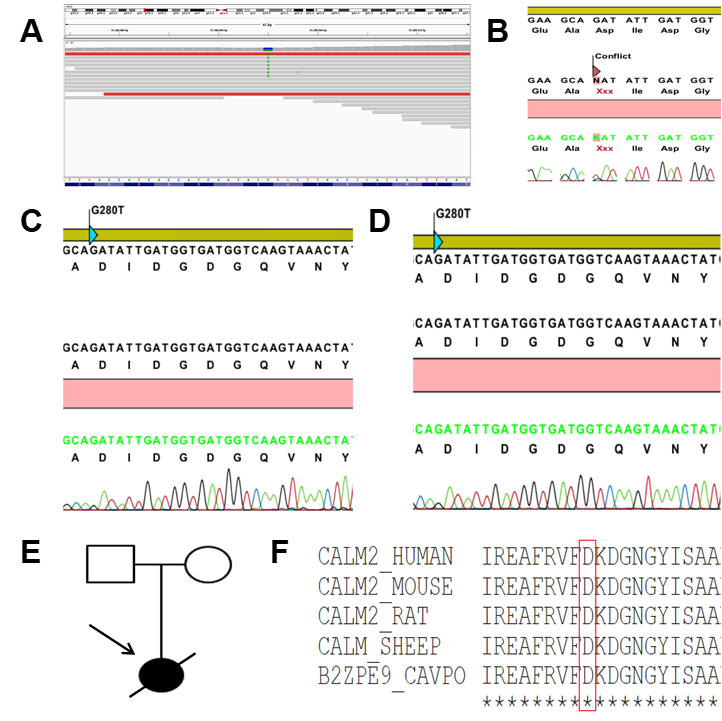
Revealing mutation in the CALM2 gene. A) Nucleotide-level sequence by the BAM file, which is the DNA reverse-complement counterpart showing a previously described missense mutation in the
To investigate the predicted impact on the calmodulin protein, the 2015 ACMG criteria, as well as widely used software scores, were taken into account. The five protein function prediction bioinformatics tools chosen were DANN, GERP, MutationTaster, PROVEAN, and SIFT. These software programs classified this variant as likely pathogenic (
To assess the possibility that the mutation c.280G > T (p.Asp94Tyr) caused SCD in the proband, WES was also performed on blood samples of the healthy parents (data not shown). Both parents were asymptomatic and did not show phenotypic abnormalities on the baseline or stress test ECGs. Additionally, they both showed a normal resting heart rate in sinus rhythm and normal exercise-induced tachycardia (data not shown). Therefore, we assumed that the mutation was not present in the patient's parents and that the mutation c.280G > T (p.Asp94Tyr) in the CaM was a cause of the SCD in the patient. Consistent with the high-throughput sequencing, the results of the Sanger sequencing revealed that the patient carried a point mutation changing aspartic acid residue (Figure 3B) while her parents did not (Figure 3C and D). The clinical status was also indicated by the family pedigree of the patient (Figure 3E).
The novel mutation c.280G > T (p.Asp94Tyr) altered the highly conserved aspartic acid residue that directly chelates Caions in EF-hand domain 3 at position Y in the pentagonal bipyramidal coordination sphere21, predicting a high pathogenic effect. Additionally, the correlation between the patient’s symptoms and the known involvement of CaM in the heart [1,22] showed the effect of the mutation on the protein function. Lastly, c.280G > T (p.Asp94Tyr) affects a highly conserved aspartic acid at position 94. This amino acid mutation point is naturally conservative. Moreover, a comparison of the protein sequence encoded by the gene of humans with that of other species indicated that the amino acid of the mutation was not changed (Figure 3F).
Discussion
In this study, we attempted to elucidate the genetic basis for LQTS with overlapping features of other CVDs. We evaluated the effectiveness of a new comprehensive NGS CM gene panel assay on a newborn index patient who suffered from CM and demonstrated that it detected mutations with high sensitivity and precision. The technique assisted in establishing a diagnosis in a complex case and amended a diagnosis based solely on clinical features.
The results showed that WES identified a novel heterozygous missense mutation in the gene. This gene is located in a CM susceptibility region (chr2p21) and encodes the CaM protein (phosphorylase kinase, delta), which is an essential signal-transducing protein that regulates calcium-dependent intracellular processes22, 23, 24, 25. As a calcium sensor protein, calcium-induced CaM modulates the function of cardiac ion channels, including the Ca1.2 calcium channel associated with LQTS, the ryanodine receptor (RyR2) associated with catecholaminergic polymorphic ventricular tachycardia (CPVT), and the Na1.5 sodium channel26, 27, 28. Importantly, three CaM genes, including , , and , which have unique nucleotide sequences that all encode for a 100% identical CaM, are expressed differently in the human heart6, 29, 30. Moreover, the pathogenic variants in these genes have all been associated with early-onset LQTS6, 13, 20, 22, 31, 32, 33, 34, 35, 36. The disease pathogenesis of calmodulinopathic LQTSs depends on (1) the severity of the calcium-binding reduction in the Ca affinity conferred by mutant CaMs, as the LQTS-causative variants reduce CaM affinity to Ca and alter the properties of the cardiac L-type calcium channel (CaV1.2), and (2) the impairment of mutant CaMs in the modulation of the Na1.5 and RyR21, 22.
Recent advances in WES have shown that approximately 10% of sporadic severe cases of CCDs are caused by mutations37, 38. The most recent studies have suggested that mutations in the gene lead to severe cardiac arrest in childhood6, 13, 31, 36. Three unrelated children aged 4 and 7 had LQTS and CPVT and carried the p.Asn98Ser mutation13, 31. An 11-year-old girl who had a p.Asp132Gly mutation was reported to suffer from LQTS, sinus bradycardia, and right bundle branch block36. A 3-week-old baby girl who had a p.Asp96Val mutation showed markedly prolonged QTc and cardiac arrest due to multiple episodes of ventricular fibrillation (VF)6. The patient included in the present study who carried the p.D94Y mutation and presented with LQTS together with TdP can be added to this list.
The genetic test results of these young patients were suggestive of a hidden channelopathy as well as a molecular diagnosis of calmodulinopathies. Surprisingly, parental genetic screening in various of the patients’ families demonstrated that calmodulinopathy-causative mutations mostly arose rather than being inherited20. Moreover, statistical studies showed that these mutations were single-nucleotide substitutions which led to distinct missense amino acid changes and were mainly distributed in the and genes20. The most important observation is that most of the calmodulinopathy-causative mutations were localized in CaM’s C-terminal Ca-binding EF-hands 3 and 4 domains, in the specific residues directly involved in Ca binding20 and predicted a high pathogenic effect. Interestingly, recent studies have shown that the distributions of the mutations in EF-hand 3 motifs, including p.Asp94Ala, p.Asp96His, p.Asp96Val, p.Asn98Ser, p.Asn98Ile, p.Ala103Val, p.Glu105Ala and p.Asp94Tyr (from the present study) all lead to LQTS or other CVDs6, 13, 32, 34, 39, 40, 41, 42.
The findings of the present study reinforce the genetic etiology for LQTS as well as SCD during infancy. Given that mutations in the CaM EF-hands 3 and 4 domains are life-threatening (since these regions directly transduce Casignals to influence the activity of ion channels, kinases and other target proteins that are essential for cardiac function), a single mutation in the C-domain can reduce the Ca binding affinity up to 53-fold6. Therefore, genes must be screened in congenital LQTS and other CVD probands because timely management may prevent SCD.
Conclusions
The present study reports on the family of a newborn girl with multiple clinical cardiac presentations who was diagnosed with LQTS type 3 (the patient carried a heterozygous nonsynonymous mutation, c.280G > T (p.Asp94Tyr), in the gene. This report reinforces the genetic etiology for LQTS and highlights the need for screening of the entire exome in congenital cardiovascular probands.
Abbreviations
LQTS: Long QT syndrome; NGS: next-generation sequencing; WES: whole exome sequencing; SCD: sudden cardiac death
Acknowledgments
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106-YS.01-2016.39.
We would also like to thank Truong Nhat Vi and Nguyen Minh Tri Viet for the contribution in this work.
Author’s contributions
Nguyen Thi Huynh Nga and Nguyen Minh Hiep wrote the manuscript. Bui Chi Bao, Nguyen Thi Huynh Nga and Nguyen Minh Hiep designed the study, analyzed and interpreted the data. Bui Chi Bao, Nguyen Vuong Thao Vy, Nguyen Thanh Tung Vu, Nguyen Manh Cong performed the experiments. All authors approved the final manuscript.
Funding
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 106-YS.01-2016.39.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Ethics approval and consent to participate
Procedures were reviewed and approved by the Ethical Committee of the University of Medicine and Pharmacy, Ho Chi Minh, Vietnam. Participant’s parents were consenting to including the research methods involved.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.