

Copyrights: Vuong Van Dang, Dung Hoang Nguyen, Si Van Nguyen, 2022. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Protein S is a glycoprotein essential in the regulation of blood coagulation. Familial protein S deficiency increases the risk of venous thromboembolism approximately 2- to 11-fold. Herein, we report this disorder in six members of a Vietnamese family among which three had thromboembolism, and the other three were asymptomatic. The protein S levels ranged from 10.1% to 24%, but we did not identify any PROS1 mutation. In one patient, a rare combination of protein C and S deficiency withPROC heterozygous c.565C>T (p.Arg189Trp) mutation was confirmed. Direct oral coagulation was predominantly selected for both treatment and prophylaxis, which yielded no thromboembolic and hemorrhagic events during long-term follow-up. The patients without overt clinical manifestations and those with minimized risk factors could be safely left untreated. The present family pedigree systematically illustrated a diagnostic approach and an integrated consideration for deploying various individualized treatment options.
Introduction
Protein S (PS) is a vitamin K-dependent glycoprotein that plays an essential role in the regulation of blood coagulation. Typically, the average plasma free protein S (FPS) level is 40% of the total plasma PS level, whereas the remaining 60% is bound to C4b-binding protein. In addition to serving as a cofactor of activated protein C (PC) in the deactivation of activated coagulation factors V and VIII, PS is also identified as a cofactor of tissue factor pathway inhibitor in the inactivation of activated coagulation factor X1.
Protein S deficiency (PSD) can be inherited in an autosomal dominant fashion or acquired as a result of several conditions, including pregnancy, vitamin K deficiency, oral contraceptive use, severe hepatic dysfunction, and chronic infections1. This familial disorder is rare, affecting only 0.03 to 0.13% of the general population and 1 to 13% of patients with venous thromboembolism (VTE)2. The risk of VTE is approximately 2- to 11-fold higher in patients with such deficiency than in healthy individuals3.
The diagnosis and management of familial PSD remain challenging and controversial. Thus, we report a pedigree with thromboembolic events associated with PSD to elucidate the diagnosis and individualized treatment of this disorder. Management involved pharmacological intervention, control of modifiable risk factors, and timely detection of thromboembolic events and hemorrhagic side effects of anticoagulants as well as mortality within a 3-year follow-up.
CASE PRESENTATION
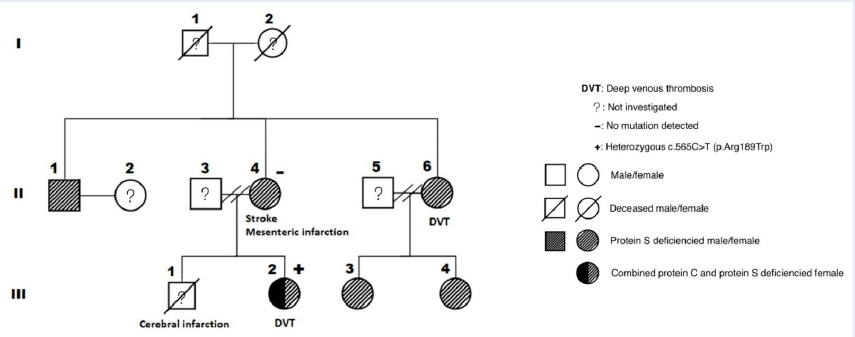
The first patient (II-4) was a 59-year-old woman (weight: 71 kg, height: 158 cm) with a history of hyperlipidemia and ischemic stroke. She had a son (III-1) who died at the age of 23 years after the onset of an acute massive cerebral infarction without any documented cardiovascular risk factors. In May 2019, she was diagnosed with mesenteric venous thrombosis, causing segmental small intestinal necrosis, which required surgical removal. The patient was then managed with anticoagulation therapy and carefully assessed for hypercoagulability, including cancer screening and comprehensive thrombophilia tests. No malignancy was detected, but the PS level was significantly reduced. The other test results are illustrated in Table 1. After several weeks of vitamin K antagonist (VKA) treatment without achieving a stable therapeutic international normalized ratio (INR), a direct oral anticoagulant (DOAC)—rivaroxaban—was prescribed and administered until the time of writing.

| Results | Normal values | |
|---|---|---|
| PT | 11.3 | 10 – 13 |
| INR | 1.03 | 1 – 1.2 |
| APTT | 33.8 | 26 – 37s |
| APTT (R) | 1.15 | 0.8 – 1.2 |
| Protein S (Free) | 24 | 62 – 145% |
| Protein C | 98.4 | 70 – 135% |
| Antithrombin III | 72.2 | 80 – 130% |
| ANA (IBL) | NEG 0.027/0.199 | |
| Anti – dsDNA | 1.31 | < 25 IU/mL |
| Lupus Anticoagulant Screen | 30.6 | 31.0 – 44.0s |
| Lupus Anticoagulant Screen Ratio | 0.94 | 0.9 – 1.1 R |
| Anti – Cardiolipin IgM | 1.5 | ≤ 20 U/mL |
| Anti – Cardiolipin IgG | 4.2 | ≤ 20 U/mL |
| Paroxysmal nocturnal hemoglobinuria (PNH) CD panel: CD55, CD59, CD14, CD24, FLAER | NEG |
| II-4 | II-6 | II-1 | III-2 | III-3 | III-4 | Normal values | |
|---|---|---|---|---|---|---|---|
| Protein S (Free) | 24 | 17.7 | 13.5 | 10.1 | 23 | 17 | 62 – 145% |
| Protein C | 98.4 | 126.0 | 82 | 61 | 121 | 127 | 70 – 135% |
| AntiThrombin III | 72.2 | 126.4 | 112.6 | 88 | 95 | 99 | 80 – 130% |
| Patient | Gene | Genotype | Heterozygou/Homozygous | Locus | Nucleotide/ Protein | Consequences | Phenotype | Group |
|---|---|---|---|---|---|---|---|---|
| II-4 | - | - | - | - | - | - | - | - |
| III-2 | PROC | Dominant/ recessive | Heterozygous | chr2: 127426114 | c.565C>T (p.Arg189Trp) | A missense mutation | Hereditary thrombophilia due to protein C deficiency | No consensus on the evidence for the disease |
One year later, her daughter (III-2), a 30-year-old cashier (weight: 78 kg, height: 163 cm), presented with a swollen right calf due to deep vein thrombosis (DVT). She was treated with rivaroxaban for 9 months; the treatment was stopped after better lifestyle modifications were achieved, yielding a weight of 66 kg. Meanwhile, the first patient’s elder sister (II-6), a 67-year-old woman (weight: 60 kg, height: 155 cm) who had a history of hypertension, hyperlipidemia, and chronic venous insufficiency, also developed DVT in her right calf. She was managed with dabigatran for 4 months, which was switched to VKA for 6 months.
Patients III-2 and II-6 also had reduced PS levels, with patient III-2 showing an additional decrease in the PC level. Consequently, other family members (II-1, III-3, and III-4) without obvious VTE were consulted and evaluated for these disorders. As expected, PSD was observed in all members (Figure 1 and Table 2).
To establish the inherited etiology, we screened patients II-4 and III-2 for mutations via next-generation sequencing (NGS). No PROS1 mutation was detected in both patients; in contrast, PROC heterozygous c.565C>T (p.Arg189Trp) mutation was identified in patient III-2 (Table 3).
DISCUSSION
Although familial thrombophilia is known to increase the risk of thrombotic events, the role of hypercoagulability is controversial. It is essential to screen individuals with suspected familial thrombophilia, including VTE, at a young age (40 to 50 years), a strong family history of VTE, unprovoked VTE, recurrent VTE events, and thrombosis in an unusual site (central nervous system or splanchnic veins)4. Based on the presence of a negative result of malignancy screening, a familial history of thrombosis, and recurrent VTE events, patient II-4 was highly suspected of having familial thrombophilia.
The screening parameters included antithrombin activation, PC deficiency and PSD, antiphospholipid syndrome, myeloproliferative neoplasm, and paroxysmal nocturnal hemoglobinuria4. Since factor V Leiden and prothrombin gene mutations are generally rare in Vietnamese and other Asian populations, these parameters are not routinely evaluated5, 6. A PS assay was then indicated in the other family members in this study, including those with pre-existing VTE and healthy individuals in which an FPS antigen assay was used as the initial choice7. Very low FPS levels in all tests generally suggest the presence of an inherited PSD.
Therefore, gene sequencing is necessary for genetic diagnosis. PS is encoded by PROS1, located in chromosome 3. More than 200 different PROS1 mutations have been identified to be associated with loss of PS function1. In this report, NGS was performed in patients II-4 and III-2 but showed no PROS1 mutation. Previous reports also showed that such mutation may not be detected in up to 50% of PSD cases1, 3. The possible causes are the unique genomic architectures of PROS1 underlying a large intragenic tandem duplication mutation or larger deletions or duplications covering the entire or a part of this gene. These mutations are difficult to characterize using a common sequencing approach. Another explanation for the absence of PROS1 mutation could be that the promoter and introns regions of PROS1 are largely unidentified and further complicated by the existence of the homologous PROS2 pseudo gene8, 9. Thus, multiplex ligation-dependent probe amplification could be used for the detection of these mutations and third-generation sequencing for the identification of other mutations outside the coding regions of PROS1. However, third-generation sequencing was not available in our facility10. Accordingly, genetic analysis was not performed among the remaining members. An interesting finding in this pedigree was the coexistence of PC deficiency and PROC heterozygous c.565C>T (p.Arg189Trp) mutation in patient III-2. This PROC mutation is also the most common mutation reported among Vietnamese patients with DVT with a low PC level11. The combined deficiency of PC and PS is rare, with a frequency of roughly 0.88% among patients with VTE, and is associated with an increased risk of thrombosis12, 13, 14. The development of VTE at a young age may be explained by the combination of both PSD and PC deficiency. Nevertheless, studies on the management of affected patients are limited.
The initial management of acute VTE in patients with familial PSD does not differ from that in individuals without this disorder15. The main agents previously used included traditional anticoagulants such as heparin (low-molecular-weight or intravenous unfractionated heparin) for in-hospital treatment, followed by warfarin. However, the competitive inhibition of warfarin by vitamin K epoxide reductase complex 1, which can deplete functional vitamin K reserves, also suppresses the synthesis of vitamin K-dependent anticoagulation factors, including PS16. This can exacerbate critical thrombotic conditions.
The recent introduction of DOACs represents a significant progress in anticoagulation. Multiple clinical trials comparing rivaroxaban and dabigatran with warfarin highlighted the effectiveness of DOACs in treating and preventing recurrent VTE as well as in decreasing bleeding complication risks17. Serrao et al. did not observe any benefits in 18 patients with PSD based on the development of hemorrhagic or thromboembolic complications within 29 months of DOAC therapy (administered front-line or after VKA)18. Yamazaki et al. illustrated that warfarin decreased the vitamin K-dependent PC activity (from 100 to 62%) and PS level (from 74 to 46%); on the contrary, edoxaban significantly increased the PC activity (from 88 to 93%), while maintaining the PS level (from 89 to 90%)19. Nevertheless, the results of the treatment efficacy for familial thrombophilia due to PSD remain controversial20.
In patient II-4, VKA was switched to DOAC because of the fluctuating INR. Not only anticoagulation therapy for 6 months but also indefinite prophylaxis should be recommended owing to the presence of both familial PSD and rare thrombosis at unusual sites.
Patient III-2 had DVT along with modifiable risk factors, including obesity, standing and sedentary work, and combined deficiency of PC and PS. Thus, in addition to DOAC therapy for 6 months, she was also encouraged to change her lifestyle, including increased physical activity for weight loss. After 9 months, she consented to DOAC cessation owing to her desire to conceive and was instructed to monitor for symptoms of thrombosis, since her risk factors were successfully minimized. No hemorrhagic or thromboembolic complications were reported thereafter.
Patient II-6 had a medical history of hypertension and hyperlipidemia and also developed DVT. She was managed with DOACs in the first 4 months and VKA in the following months owing to financial issues. Thereafter, anticoagulation was stopped on demand. Detailed lifestyle modification and self-monitoring instructions were provided. Her total duration of anticoagulation was 10 months, and thrombosis has not recurred to date.
Patients II-1, III-3, and III-4, who had no thrombotic manifestations, did not receive anticoagulation treatment. They were only instructed to recognize the symptoms of thromboembolism and practice a better lifestyle21.
Women of childbearing age with PSD should be more knowledgeable about such disorder, as they are at a very high risk of antenatal and postpartum VTE (antepartum: 0.9%, postpartum: 4.2%)22. Hormone use is associated with an increased risk of thrombosis in these individuals23. Herein, patients III-2, III-3, and III-4 were provided with necessary information to avoid unwanted circumstances.
The choice of management was dependent on the clinical manifestations and individual risk factors. We considered the individual cases to provide effective management, increase adherence, and reduce the possibility of over-management, which yields more harm than good. This real-life approach was proven to be effective and safe for all members within the 3-year follow-up in this study.
This report has some limitations. First, the genetic testing technique used in the study might not be reliable enough to detect variant mutations occurring outside coding regions, large deletions, duplication or insertion (over 100 nucleotides) mutations, short tandem repeats, and pseudogenes (PROS2)8. Although the culprit mutation could not be found, the technique did not interfere with the offspring screening via PS level measurement. Second, the patients did not receive the same anticoagulant owing to the high cost of treatment; nevertheless, this illustrates the real-life practice in developing countries.
CONCLUSIONS
Our report systematically presented a diagnostic approach including necessary tests and an integrated consideration for deploying various individualized treatment options. Notably, the presence of PSD should be evaluated among family members suspected of having inherited this condition. PSD and PC deficiency could co-exist although very rarely. Finally, DOACs are considered an appropriate choice for both treatment and prophylaxis because of their effectiveness and low risk of complications, although the high treatment cost is an important interfering factor.
Abbreviations
None.
Acknowledgments
None.
Author’s contributions
VVD: Conceived the study, acquired patients’ information and drafted the manuscript; DHN: Participated in the study design and coordination, and drafted the manuscript; SVN: Conceived the study, and critically revised the manuscript. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
This study was conducted in accordance with the amended Declaration of Helsinki. Written informed consent was obtained from the patient for publication of this Case Report and any accompanying images.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Kate
M. K. Ten,
Meer
J. van der,
Protein S deficiency: a clinical perspective. Haemophilia : the official journal of the World Federation of Hemophilia.
2008;
14
(61)
:
1222-1228
.
View Article Google Scholar -
Dykes
A.C.,
Walker
I.D.,
McMahon
A.D.,
Islam
S.I.,
Tait
R.C.,
A study of Protein S antigen levels in 3788 healthy volunteers: influence of age, sex and hormone use, and estimate for prevalence of deficiency state. British Journal of Haematology.
2001;
113
(3)
:
636-41
.
View Article PubMed Google Scholar -
Wypasek
E.,
Undas
A.,
Protein C and protein S deficiency - practical diagnostic issues. Advances in clinical and experimental medicine : official organ Wroclaw Medical University.
2013;
22
(4)
:
459-467
.
-
Connors
J.M.,
Thrombophilia Testing and Venous Thrombosis. The New England Journal of Medicine.
2017;
377
(12)
:
1177-87
.
View Article PubMed Google Scholar -
Angchaisuksiri
P.,
Venous thromboembolism in Asia: an unrecognised and under-treated problem?. Thrombosis and Haemostasis.
2011;
106
(4)
:
585-90
.
View Article PubMed Google Scholar -
Duc Bach
N.,
Anh Vu
H.,
Van Dung
P.,
Hoai Nam
N.,
Factor
V.,
Leiden in patients with deep venous thrombosis. Ho Chi Minh J Med..
2020;
18
:
124-9
.
-
Marlar
R.A.,
Gausman
J.N.,
Tsuda
H.,
Rollins-Raval
M.A.,
Brinkman
H.J.,
Recommendations for clinical laboratory testing for protein S deficiency: communication from the SSC committee plasma coagulation inhibitors of the ISTH. Journal of Thrombosis and Haemostasis.
2021;
19
(1)
:
68-74
.
View Article PubMed Google Scholar -
Lanke
E.,
Johansson
A.M.,
Hillarp
A.,
Lethagen
S.,
Zöller
B.,
Dahlbäck
B.,
Co-segregation of the PROS1 locus and protein S deficiency in families having no detectable mutations in PROS1. Journal of Thrombosis and Haemostasis.
2004;
2
(11)
:
1918-23
.
View Article PubMed Google Scholar -
Seo
J.Y.,
Lee
K.O.,
Kim
S.H.,
Oh
D.,
Kim
D.K.,
Kim
H.J.,
The genomic architecture of the PROS1 gene underlying large tandem duplication mutation that causes thrombophilia from hereditary protein S deficiency. Gene.
2014;
547
(2)
:
295-9
.
View Article PubMed Google Scholar -
Pintao
M.C.,
Garcia
A.A.,
Borgel
D.,
Alhenc-Gelas
M.,
Spek
C.A.,
de Visser
M.C.,
Gross deletions/duplications in PROS1 are relatively common in point mutation-negative hereditary protein S deficiency. Human Genetics.
2009;
126
(3)
:
449-56
.
View Article PubMed Google Scholar -
Do
MD,
Pham
DV,
Le
LP,
Le
LH Gia,
Recurrent PROC and novel PROS1 mutations in Vietnamese patients diagnosed with idiopathic deep venous thrombosis. International journal of laboratory hematology.
2021;
43
(2)
:
266-272
.
View Article Google Scholar -
Suehisa
E.,
Nomura
T.,
Kawasaki
T.,
Kanakura
Y.,
Frequency of natural coagulation inhibitor (antithrombin III, protein C and protein S) deficiencies in Japanese patients with spontaneous deep vein thrombosis. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis.
2001;
12
(2)
:
95-99
.
View Article Google Scholar -
Patel
M.L.,
Sachan
R.,
Seth
G.,
Combined deficiency of proteins C and S: ischaemic stroke in young individuals. BMJ Case Reports.
2013;
2013
.
View Article PubMed Google Scholar -
Hayashida
M.,
Yamada
H.,
Yamazaki
S.,
Nomura
H.,
Yoshimura
K.,
Kitahara
O.,
Combined protein C and protein S deficiency in a family with repetitive thromboembolism and segregated gene mutations. Internal Medicine (Tokyo, Japan).
2003;
42
(3)
:
268-72
.
View Article PubMed Google Scholar -
Khan
S.,
Dickerman
J.D.,
Hereditary thrombophilia. Thrombosis Journal.
2006;
4
(1)
:
15
.
View Article PubMed Google Scholar -
Patel
S.,
Singh
R.,
Preuss
C.V.,
Patel
N.,
Warfarin Internet: StatPearls 2021 [updated February 17, 2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470313/.. 2021
.
-
Goldhaber
Z.,
Eriksson
H.,
Kakkar
A.,
Schellong
S.,
Feuring
M.,
Fraessdorf
M.,
Efficacy of dabigatran versus warfarin in patients with acute venous thromboembolism in the presence of thrombophilia: Findings from RE-COVER®, RE-COVER™ II, and RE-MEDY™. Vascular medicine (London, England).
2016;
21
(6)
:
506-514
.
View Article Google Scholar -
Serrao
A.,
Lucani
B.,
Mansour
D.,
Ferretti
A.,
Baldacci
E.,
Santoro
C.,
Direct Oral Anticoagulants in Patients Affected by Major Congenital Thrombophilia. Mediterranean Journal of Hematology and Infectious Diseases.
2019;
11
(1)
:
e2019044
.
View Article PubMed Google Scholar -
Yamazaki
H.,
Yagi
S.,
Torii
Y.,
Amano
R.,
Oomichi
Y.,
Sangawa
T.,
Edoxaban improves acute venous thromboembolism while preserving protein C and protein S levels. Journal of Cardiology.
2018;
71
(3)
:
305-9
.
View Article PubMed Google Scholar -
Undas
A.,
Góralczyk
T.,
Direct Oral Anticoagulants in Patients with Thrombophilia: Challenges in Diagnostic Evaluation and Treatment. Advances in clinical and experimental medicine : official organ Wroclaw Medical University.
2016;
25
(6)
:
1321-1330
.
View Article Google Scholar -
Gupta
A.,
Tun
A.M.,
Gupta
K.,
Tuma
F.,
Protein S Deficiency Internet: StatPearls; 2020 [updated September 13, 2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK544344/. 2020
.
-
Croles
F.N.,
Nasserinejad
K.,
Duvekot
J.J.,
Kruip
M.J.,
Meijer
K.,
Leebeek
F.W.,
Pregnancy, thrombophilia, and the risk of a first venous thrombosis: systematic review and bayesian meta-analysis. BMJ (Clinical Research Ed.).
2017;
359
:
j4452
.
View Article PubMed Google Scholar -
Lipe
B.,
Ornstein
D.L.,
Deficiencies of natural anticoagulants, protein C, protein S, and antithrombin. Circulation.
2011;
124
(14)
:
e365-8
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 9 No 10 (2022)
Page No.: 5326-5331
Published on: 2022-10-29
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 3690 times
- PDF downloaded - 1306 times
- XML downloaded - 0 times
