

Copyrights: Jing Yit Pua, Zamzuri Idris, Abdul Aziz Mohamed Yusoff, Azim Patar, 2022. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Isocitrate dehydrogenase (IDH) mutations have been the focus of glioma-related neuroscience research since the discovery of the gene in 2008. IDH1 has been identified as a key enzyme in cellular metabolism, epigenetic regulation, redox regulation, and DNA repair and is thought to be one of the most important factors in gliomagenesis. IDH1 mutations cause neomorphic activity, resulting in an increase in 2-hydroxyglutarate production and a decrease in NADPH production. Emerging research has identified IDH1 mutations in the vast majority of low-grade gliomas and secondary glioblastomas, but these mutations are extremely uncommon in primary glioblastomas. Other genetic defects appear to play a significant role in glioma initiation and progression. In this study, we review recent findings on oncogenic alterations in IDH1, TP53, and CASP9 to identify any potential molecular correlations and interrelationships that lead to gliomagenesis. The roles and molecular interactions of these glioma-associated genes in gliomagenesis are elucidated. In addition, we highlight studies on stem cell modeling in glioma-associated genetic alterations that have been conducted over the past several decades.
Introduction
Gliomas have been identified as the most prevalent primary cancer of the central nervous system (CNS) in humans. Initially, glioma classifications were based on specific histologic subtypes, with astrocytomas, ependymomas, and oligodendrogliomas being the most prevalent, followed by brainstem, optic nerve, and mixed gliomas. Glioblastoma is the most invasive, aggressive, and lethal type of glioma; this is classified as a high-grade glioma (HGG) (World Health Organization [WHO] grade IV) and has a poor prognosis1. The Cancer Genome Atlas (TCGA) identifies four subtypes of glioma based on their predominant genetic or epigenetic alterations in gene expression: 1) proneural, 2) neural, 3) mesenchymal, and 4) classical2, 3. For instance, primary glioblastoma can be classified in any of the subtypes, whereas secondary glioblastoma is always classified as proneural. The WHO categorizes gliomas as low-grade gliomas (LGG; grades I and II) and HGGs (grades III and IV). LGGs are typically well-differentiated and slow-growing tumors, whereas HGGs are less differentiated, anaplastic, or diffusive tumors that infiltrate the brain parenchyma, thus making surgical resection challenging4.
The discovery of isocitrate dehydrogenase isoform 1 (IDH1) mutations in gliomas has prompted extensive research into their direct and indirect roles in gliomagenesis5. IDH1 remains the benchmark for the subtype classification of gliomas. Its mutation is frequently associated with TP53 mutations in astrocytic tumors, but these tumors rarely exhibit co-deletion of chromosomes 1p and 19q, which is more prevalent in oligodendrogliomas6, 7, 8. TP53 mutations are uncommonly associated with oligodendrogliomas7, while mutations in the capicua gene6 are more common. Figure 1 depicts a schematic summary of gliomagenesis based on the IDH1 mutation status.
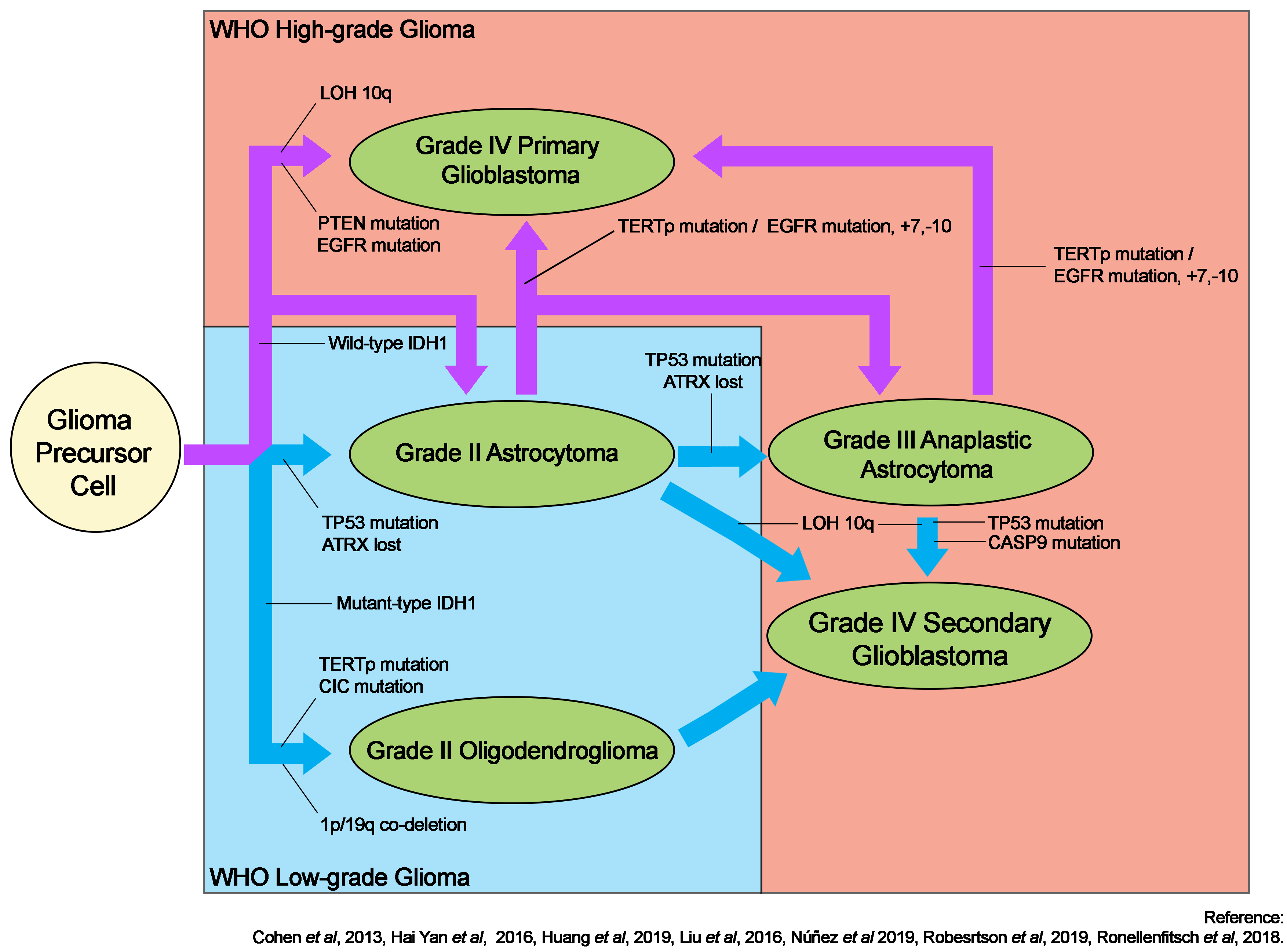
Although IDH1 mutations are frequently observed in gliomas, they are uncommon in primary glioblastomas. Glioblastoma typically develops through a variety of pathways. Primary glioblastoma develops de novo, with or without lower-grade precursors, and acquires multiple complex genetic alterations in epidermal growth factor receptor (EGFR) and phosphatase and tensin homolog (PTEN)6, 7. A mutation in the telomerase reverse transcriptase promoter has been linked with glioblastoma, in particular primary glioblastoma arising from astrocytic glioma, and grade II oligodendroglioma9. Secondary glioblastoma may also arise from grade II or III astrocytomas; this is frequently associated with loss of heterozygosity of chromosome 10q10 but not chromosomal mutation +7/10 in the context of EGFR mutations typically found in astrocytic glioblastoma11. Oligodendrogliomas may subsequently transform into anaplastic oligodendroglioma or secondary glioblastoma12.
In the United States, glioma is the most prevalent cancer among children and young adults aged 20 to 39 years13, 14. According to the National Cancer Institute’s Surveillance, Epidemiology, and End Results Program, there were approximately 24,000 new cases of brain and other nervous system cancers in the United States in 2019, accounting for 1.4% of all new cancer cases15. In Malaysia, leukemia and lymphoma are surpassed by brain and CNS tumors as the third most prevalent cancer in children16. In accordance with Sustainable Development Goal 3 of the United Nations (good health and well-being), this global health issue warrants increased focus.
In 2016, the WHO revised its classification system for brain and spinal cord tumors by incorporating key genetic alterations into the classification17, 18. Previously, the classification of primary and secondary glioblastomas was based solely on clinical and histopathological presentations; however, this method is subjective, resulting in misclassification. Histopathologically, high-grade tumors are indistinguishable6. Further, a number of alterations in genes, including IDH16, 8, 1p/19q co-deletion7, TP531, CASP919, PTEN7, cyclin-dependent kinase inhibitor 2A2, and EGFR3, are associated with gliomas. This review focuses on IDH1, TP53, and CASP9 as potential drivers of gliomagenesis and seeks to identify potential correlations between these genes in gliomagenesis.
Molecular alterations in IDH1, TP53, and CASP9 in gliomagenesis
IDH1 mutations were first discovered in LGGs but are also frequently found in HGGs5, 6, 7, 8, 20. Numerous studies have demonstrated a significant correlation between IDH1 and various histological tumor types, particularly secondary glioblastoma6, 7, 8. Over 80% – 90% of IDH1 mutations in gliomas are missense mutations that replace arginine (CGT) with histidine (CAT) at nucleotide 395 and codon 132 in exon 4 (IDH1R132H, c.395 G>A)6, 7, 8, 16, 20, 21, 22. Less common polymorphisms at codon 132 include IDH1R132C, IDH1R132L, IDH1R132S, IDH1R132G, IDH1R132V, and IDH1R132P 7, 8, 9, 10, 11, 20, 21, 22, 23. Interestingly, IDH1R132C has been associated with Maffucci syndrome24, while IDH1R132L has not been observed in immunohistochemistry23. Non-IDH1R132H mutations were identified in DNA analyses but not in immunohistochemistry25. Non-IDH1R132H mutations may be unique to secondary glioblastoma but evade detection in early LGG23, 25. Recently, IDH1 mutations have been reported to acquire chemoresistance26 and drug resistance27. Therefore, we strongly recommend incorporating molecular diagnostics into clinical settings, particularly for patients with glioma, to detect the IDH1 mutation status.
The p53 protein encoded by the TP53 gene plays a crucial role as the hub of the cellular regulatory network in regulating cell proliferation, senescence, apoptosis, genome integrity, and other regulatory functions28, 29, 30. Notably, TP53 gene mutations are one of the most prevalent biomarkers associated with gliomas, as they have been identified in virtually all cancers. Surprisingly, Yan et al. demonstrated that TP53 mutations were more prevalent in certain subtypes of gliomas such as diffuse astrocytomas, anaplastic astrocytomas, and secondary glioblastomas, which were also strongly associated with IDH1 mutations8; meanwhile, TP53 mutations were less commonly found in oligodendrogliomas or anaplastic oligodendrogliomas28, 29, 31. In addition, these tumors demonstrated 1p/19q co-deletion less frequently than did oligodendrogliomas, which yielded 1p/19q co-deletion almost universally, despite possibly harboring TP53 mutations1. The reported TP53 hotspot mutations in gliomas include S127P [nucleotide 379; serine (TCC) to proline (CCC)], R175H [nucleotide 524, arginine (CGC) to histidine (CAC)], G245S [nucleotide 733, glycine (GGC) to serine (AGC)], R248Q [nucleotide 743, arginine (CGG) to glutamine (CAG)], S260A [nucleotide 778, serine (TCC) to alanine (GCC)], R273H [nucleotide 818, arginine (CGT) to histidine (CAT)], and R273Y [nucleotides 817 and 818, arginine (CGT) to tyrosine (TAT)] (32). Kawasoe et al. reported that TP53 mutation was found in the early development of astrocytoma32. In contrast, TP53 mutations and 1p/19q co-deletion are nearly mutually exclusive.
Although germline mutations were rarely reported, somatic mutations account for many brain tumors. This is attributed to the fact that most gliomas are sporadic and have no known predisposing germline variants30. Since it was recently reported as a germline mutation found in patients with glioma, CASP9 gene mutation has garnered considerable research interest19. CASP9 mutations include R65X [nucleotide 193, arginine (CGA) to a stop codon (TGA)] and Q221R [nucleotide 662, glutamine (CAG) to arginine (CGG)]19, 33. The caspase-9 enzyme encoded by the CASP9 gene is a key biomolecule in the p53-dependent mitochondrial programmed cell death pathway. We hypothesized that CASP9 mutation and TP53-mediated gliomagenesis are closely related.
Molecular etiology of gliomas based on IDH1, TP53, and CASP9
IDH1 mutation causes profound changes at the cellular level, including alterations in cellular metabolism. IDH1 gain-of-function mutations reduce -ketoglutarate (-KG) to the oncometabolite (R)-enantiomer of 2-hydroxyglutarate (R-2-HG)34. Evaluation of clinical and cultured samples revealed elevated 2-HG levels in glioma cells harboring IDH1 mutation7, 35. Eventually, the increased concentration of 2-HG reduces the production of NADPH and halts the oxidative decarboxylation of isocitrate21. IDH1R132H mutation inactivates the ability to bind isocitrate catalytically and reduces the activity of the enzyme35. Competition between 2-HG and α-KG at catalytic sites further inhibits α-KG-dependent enzyme function, rendering cells susceptible to pharmacological glutaminolysis inhibition36, 37. The α-KG-dependent enzymes include collagen prolyl-4-hydroxylase, prolyl hydroxylase, the ten–eleven translocation (TET) family of 5 methylcytosine hydroxylases, the Jumonji domain-containing family of histone lysine demethylases, enzymes involved in nucleic acid metabolism, and other enzymes with still unknown functions34. The glutaminolysis pathway has been identified as being dependent on IDH mutations, and IDH1-mutant glioma has been hypothesized to rely on glutamate rather than on glutamine38. In fact, IDH1-mutant tissues display a significant decrease in glutamine levels but not in cell cultures35. Accordingly, IDH1R132H mutation may be a driver mutation in gliomas likely through the production of 2-HG, which leads to the previously mentioned conditions. However, it remains unknown whether IDH1 mutation, 2-HG, or both promote oncogenic events.
IDH1 mutations may cause hypermethylation, gene-specific hypomethylation, and genome-wide hypomethylation, increasing G-CIMP (glioma CpG island methylator phenotype) production39. DNA methylation is essential for the regulation of gene activity and nuclear structure as well as the development and progression of cancer40, 41. CIMP hypermethylation in the promoter regions of tumor-suppressor genes has been linked with numerous cancers39. The prevalence of hypermethylation is fourfold higher in glioma than in other cancers with IDH mutations39; this indicates that gliomas with IDH mutations have distinctive molecular characteristics compared with other cancers with IDH mutations, in which significantly elevated CIMP levels are rarely observed. Liu hypothesized that IDH1 mutation and the G-CIMP phenotype are unique to gliomas22, while Kamiska claimed that G-CIMP is a prognostic factor34, with low levels being associated with poor outcomes30, 39. Similarly, loss of DNA methylation is associated with the progression of glioma7. In a neurosphere model, 2-HG is significantly associated with inhibition of lysine demethylases and the TET family, which results in hypermethylation of histone 3 and epigenetic reprogramming of the glioma transcriptome39, 40. However, changes in histone methylation lead to the suppression of cell differentiation34. CIMP may not be the actual oncogenic transformation factor, at least in astrocytes, because IDH1 mutation introduction did not result in tumor formation6. Silencing epigenetic targets may reduce glioma malignancy and yield favorable clinical outcomes.
IDH1 protects cells against reactive oxygen species (ROS) by generating GSH. NADPH is also involved in lipid metabolism and contributes to cell defense against ROS during lipid oxidation34. Its mutation reduces the pool of GSH by decreasing the levels of -KG and NADPH, an essential cofactor for maintaining normal levels of GSH, resulting in increased susceptibility to ROS38. As a result of exposure to free radicals, cells become more susceptible to oxidative damage and eventually die6. Unexpectedly, in vitro studies have demonstrated an inverse relationship between GSH and ROS in cancer cells carrying IDH1 mutations. In neither the brain nor the hematopoietic cells of IDH1 knock-in animal models nor immortalized human astrocyte cell lines were the aforementioned conditions observed38.
IDH1R132H harboring glioma tumors and cultured glioma cells have recently been found to have reduced levels of β-oxidation and carnitine. Miyata was the first to discover that oxidation decreased only in IDH-mutant gliomas owing to the decrease in carnitine levels and that the difference in carnitine levels was significantly greater than that in 2-HG levels35. The changes in carnitine levels may explain why patients with IDH1 mutations have better prognosis than those with wild-type IDH1 mutations, as reduced β-oxidation activity inhibits tumor growth by depleting ATP for cancer cell division. Miyata suggested that carnitine is a better biomarker than 2-HG for detecting IDH mutations, given that patients with mutant-type IDH1 gliomas had significantly lower levels of carnitine than those with wild-type IDH1 gliomas35. Interestingly, the decrease in carnitine levels and β-oxidation activities varied considerably between clinical samples and cell lines35. The possibility of carnitine as a better biomarker and its relationship to the prognostic value in patients with glioma must be validated.
High levels of 2-HG have been known to inhibit prolyl hydroxylase domain (PHD) and hypoxia-induced factor 1 subunit alpha (HIF1-α), leading to angiogenesis that supports the development of glioma34. Thus, gliomas harboring mutant-type IDH1 express lower levels of HIF1-α than do those harboring wild-type IDH1. HIF1-α is also a transcription factor that aids in the adaptation to hypoxic conditions, thus impacting the hypoxia status, angiogenesis, metabolism, growth and differentiation, apoptosis and autophagy, and cell motility34. Moreover, Zhao suggested that PHD enzymes are the key drivers for HIF1-α degradation and hydroxylation42. These enzymes require α-KG and ferrous iron (Fe2+) as a cofactor for the degradation of HIF1-α. Thus, IDH1 mutations that reduce the levels of α-KG may stimulate the cellular accumulation of HIF1-α7. As a result of decreased HIF1-α expression, vascular endothelial growth factor is overproduced, allowing angiogenesis and vasculogenesis critical for tumor growth and metastasis via the vasculature6, 7. Increased HIF1-α levels and gene expression were also observed in vitro in U87MG cell lines, but the effect was abolished by exogenous administration of an α-KG derivative42. In brief, IDH1 mutation induces an overproduction of 2-HG oncometabolite molecules, leading to the production of HIF1-α that supports the vascularization of tumors and allows the growth and metastasis of tumors.
IDH1 mutation can increase the sensitivity of gliomas to radiation6. However, increased survival in an animal model was only achieved with pharmacological treatment, not with radiation40. G-CIMP hypermethylation modulates glioma sensitivity to drugs and radiotherapy, which consequently enhances resistance to ionizing radiation22. This mutation induces genomic stability but reduces the efficacy of radiotherapy in animal models40; it further induces extensive DNA hypermethylation and reshaping of the methylome, mirroring G-CIMP-positive LGG in primary human astrocytes43. Accordingly, IDH1 mutation might decelerate the growth of glioma and increase DNA repair capacity, although it remains unclear whether it increases or decreases radiosensitivity. Patients with IDH1-mutated glioma generally exhibit a longer survival period than do patients with wild-type IDH1-mutated glioma, perhaps because of the effects of the mutation on G-CIMP. Almost all G-CIMP-positive tumors possess an IDH1 mutation, while no G-CIMP-negative tumors have been found to carry such mutation43. Thus, G-CIMP and IDH1 mutations are tightly associated. In the context of TP53 and ATRX inactivation, IDH1 mutation stimulates the ataxia–telangiectasia signaling pathway, consequently inducing DNA damage response and genomic stability owing to epigenetic reprogramming mechanisms involving chromatin modifications via histone lysine demethylation33, 40. Although G-CIMP is a major determinant of gliomagenesis, its molecular basis remains poorly understood.
The p53 pathway is frequently deregulated in glioblastoma. TCGA reported that the p53 pathway is deregulated in >80% of gliomas44 and up to 90% of glioblastoma cell lines37. The p53 pathway is disrupted in response to DNA damage and genotoxicity, oncogene activation, aberrant growth signaling, and hypoxia1. The particular mutations are typically missense mutations in the DNA-binding domain, abrogating transcription factor activity1. Under normal conditions, p53 ubiquitination is mediated by ubiquitin ligase in a negative feedback loop to regulate p53 protein activity. However, this is disrupted by TP53 missense mutations. In contrast to wild-type TP53, mutant-type TP53 results in loss of wild-type function and gain-of-function, causing the accumulation of p53 proteins, which then triggers enhanced proliferation, invasion, reduced chemosensitivity, carcinogenic metabolism, disturbed tissue architecture, and tumor initiation and progression1, 28. A shorter survival was observed in patients with mutant-type TP53 and IDH1, while the prognosis of patients with wild-type IDH1 was not affected1, 6. Although TP53 mutation is found in almost all cancer types, TP53 mutation status allows better understanding of the tumor biology of gliomas, especially in the context of co-occurrence with other gene mutations.
CASP9 R65X mutation causes loss of the catalytic domain of caspase-9. This mutation is predicted to have functional consequences in the p53 signaling pathway. In response to DNA damage, p53 promotes pro-apoptotic biomolecules and inhibits anti-apoptotic biomolecules, activating caspase-919. The active form of caspase-9 triggers a cascade of effector caspases, resulting in cell death. However, CASP9 mutation blocks the p53 programmed cell death cascade, allowing the survival of DNA-damaged cells. Apoptosis is defined as a physiological process of programmed cell death; thus, defects in this mechanism can lead to abnormal cell growth and proliferation. A summary of the roles of IDH1, TP53, and CASP9 gene mutations based on 2-HG production in gliomagenesis is shown in Figure 2. Additional studies are needed to examine whether IDH1, TP53, and CASP9 mutations are involved in tumor progression or result from a damaged DNA repair mechanism in tumors.

Stem cell modeling in glioma-associated genetic alterations
There are increasing efforts to manipulate the current technology of genetically modified or orthotopic induction animal models that develop malignant brain tumors. Owing to animal ethics, different assays or paradigms to enrich brain tumor biology, including that of cancer stem cells, have been used in brain tumor research45. One frequently used system involves placing primary human GBM cells in suspension with serum-free defined growth factors46. This approach has been used to study in vitro growth properties, chemosensitivity and chemoresistance, epigenetic characteristics, and gliomagenesis potential46, 47. Another system is an immunological approach in which antibody-mediated selection for putative glioma cell-specific epitopes is applied47. Examples of the well-known epitopes of glioma cells are CD133, CD44, CD15, and SSEAs47. Brain stem cells are enriched in populations by the ability to efflux the Hoechst 33342 dye. Next-generation sequencing has played an important role in deciphering the genomic landscape of gliomas, therefore leading to the era of molecular biology- and stem cell-based approaches for the study of genetic alterations in gliomas. More recently, induced pluripotent stem cell (iPSC) technology, 3D bioprinting, and 3D culture technique have been introduced to model gliomas. Herein, we describe stem cell modeling in glioma-associated genetic alterations as they have been thoroughly investigated.
The cancer stem cell theory postulates that tumors are sustainable owing to their specific features, including self-renewal ability. Neural stem cells are normally non-dividing populations but can be induced to proliferate under conditions of stress48. Llagun and Parada suggested that glioma stem cells originate from a neural stem cell; therefore, glioma and neural stem cells share common phenotypic markers46, 48. Glioma stem cells are distinguishable based on their genetic alterations. The most frequent genetic alterations found in gliomas are alterations in the genes that encode proteins implicated in signaling cascades or cell cycle control, as previously described.
Fantin and colleagues experimented on U87MG and LN-18 human glioblastoma stem cells transfected with Myc-tagged wild-type IDH1 and mutant-type IDH1R132H mutations to profile the changes in 2-HG levels between glioma stem cell models49. They found that IDH1R132H-expressing stem cells had a significantly higher level of 2-HG than wild-type IDH1-expressing stem cells49. The 2-HG level is elevated in mutant-type IDH1 tumor samples. The stem cell model could be mimicking mutant-type IDH1-specific glioma stem cells. The use of glioma stem cell modeling for investigating glioma-associated genetic alterations might be reliable and be an alternative method for animal models, supporting the 3R concept (Replacement, Reduction, Refinement) in brain tumor research. This notion was further supported by the findings by Shi et al. who used two different human glioblastoma cell lines (U87MG and U251MG) to demonstrate IDH1 gene alterations by site-directed mutagenesis and lentivirus transfection50. They found that the IDH1 mutation stem cell model showed increased chemosensitivity, decreased GSH and NADPH levels, and increased ROS production, similar to the changes found in patient tumor samples in other studies50.
Olafson et al. analyzed mutated genes from a glioma patient-derived cell line model. Two low-passage patient-derived primary cell lines were established in the laboratory and then analyzed via pyrosequencing, in situ hybridization, immunohistochemistry, and next-generation sequencing to validate gene mutations in the stem cell model28. They found PTEN, EGFR, MAP3KI, NTRKI and TP53 gene mutations in both cell line models in next-generation sequencing, although they were able to detect mutant-type TP53 gene in only one of the stem cells28. The mutational data revealed molecular profiles similar to tumors.
More advancements have been achieved by integrating iPSC technology into the study of glioma-initiating cells51 and modeling of LGGs51. Further, 3D bioprinting as a scaffold for cell culture was recently used to study the invasion ability of glioma52. The discovery of cerebral organoids53 has opened up a new research niche in the investigation of gliomas. Several studies demonstrated that 3D culture of cerebral organoids provides a superior understanding of glioma at another level54, 55, 56.
Stem cell modeling in glioma-associated genetic alterations is an essential alternative method from animal models. An increasing number of studies use stem cell models in brain tumor biology. Although the roles of several genes in glioma stem cell models remain uncertain, stem cell modeling for gliomas can reveal valuable information for brain research.
Conclusions
The metabolic and biological consequences of genetic mutations can improve the understanding of the development and clinical behavior of different molecular subtypes of glioma. However, the ability to target gliomas with IDH mutations remains limited. Thus, an in-depth understanding of the fundamental biology of gliomas is warranted to improve the knowledge of tumor biology and will be of great clinical and prognostic importance for neuroscience. Discoveries of targets and therapeutic strategies may soon emerge.
Abbreviations
2-HG: 2-hydroxyglutarate, 3R’s: replacement, reduction and refinement, ATP: adenosine triphosphate, CAG: glutamine, CASP9: Caspase-9, CAT, CAC: histidine, CCC: proline, CD133, CD44, CD15: cell of differentiation 133, 44, 15; CGT, CGC, CGG: arginine, CIMP: CpG island methylator phenotype, DNA: deoxyribonucleic acid, EGFR: epidermal growth factor receptor, GBM: glioblastoma, GCC: alanine, G-CIMP: glioma CpG island methylator phenotype, GGC: glycine, GSH: glutathione, HGG: high grade glioma, HIF1- α: hypoxia-induced factor 1 subunit alpha, IDH: Isocitrate dehydrogenease, IPSC: induced pluripotent stem cell, JmjC: Jumonji domain-containing family, LGG: low grade glioma, MAP3KI: mitogen-activated protein kinase 1, NADPH: Nicotinamide adenine dinucleotide phosphate, NTRKI: neurotropic tyrosine receptor kinase 1, PHD: prolyl hydroxylase domain, PTEN: phosphatase and tensin homolog deleted on chromosome 10, R-2-HG: R-enantiomer of 2-hydroxyglutarate, ROS: reactive oxygen species, SSEAs: stage-specific embryonic antigens, TAT: tyrosine, TCC, AGC: serine, TCGA: The Cancer Genome Atlas, TET: ten-eleven translocation, TGA: stop codon, TP53: tumor protein 53, VEGF: vascular endothelial growth factor, WHO: world health organization, α-KG: alpha-ketoglutarate
Acknowledgments
We would like to thank all the members of the laboratory of AP for fruitful discussions.
Author’s contributions
Conceptualization, JYP, ZI, AAMY, and AP Writing—original draft preparation, JYP; writing—review and editing, ZI, AAMY, and AP; supervision, AP.; funding acquisition, ZI, AAMY, and AP. All authors have read and agreed to the published version of the manuscript.
Funding
The Azim Patar laboratory is supported by the Ministry of Higher Education Malaysia for Fundamental Research Grant Scheme with Project Code FRGS/1/2019/SKK08/USM/03/10.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Zhang
Y.,
Dube
C.,
Gibert
M.,
Cruickshanks
N.,
Wang
B.,
Coughlan
M.,
The p53 pathway in glioblastoma. Cancers (Basel).
2018;
10
(9)
:
297
.
View Article PubMed Google Scholar -
Network
Cancer Genome Atlas Research,
Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature.
2008;
455
(7216)
:
1061-1068
.
View Article PubMed Google Scholar -
Verhaak
R.G.,
Hoadley
K.A.,
Purdom
E.,
Wang
V.,
Qi
Y.,
Wilkerson
M.D.,
Cancer Genome Atlas Research Network
Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell.
2010;
17
(1)
:
98-110
.
View Article PubMed Google Scholar -
de Robles
P.,
Fiest
K.M.,
Frolkis
A.D.,
Pringsheim
T.,
Atta
C.,
St Germaine-Smith
C.,
The worldwide incidence and prevalence of primary brain tumors: a systematic review and meta-analysis. Neuro-Oncology.
2015;
17
(6)
:
776-83
.
View Article PubMed Google Scholar -
Balss
J.,
Meyer
J.,
Mueller
W.,
Korshunov
A.,
Hartmann
C.,
von Deimling
A.,
Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathologica.
2008;
116
(6)
:
597-602
.
View Article PubMed Google Scholar -
Cohen
A.L.,
Holmen
S.L.,
Colman
H.,
IDH1 and IDH2 mutations in gliomas. Current Neurology and Neuroscience Reports.
2013;
13
(5)
:
345
.
View Article PubMed Google Scholar -
Huang
L.E.,
Friend or foe-IDH1 mutations in glioma 10 years on. Carcinogenesis.
2019;
40
(11)
:
1299-307
.
View Article PubMed Google Scholar -
Yan
H.,
Parsons
D.W.,
Jin
G.,
McLendon
R.,
Rasheed
B.A.,
Yuan
W.,
IDH1 and IDH2 mutations in gliomas. The New England Journal of Medicine.
2009;
360
(8)
:
765-73
.
View Article PubMed Google Scholar -
Robertson
F.L.,
Marqués-Torrejón
M.A.,
Morrison
G.M.,
Pollard
S.M.,
Experimental models and tools to tackle glioblastoma. Disease Models & Mechanisms.
2019;
12
(9)
.
View Article PubMed Google Scholar -
Soomro
S.H.,
Ting
L.R.,
Qing
Y.Y.,
Ren
M.,
Molecular biology of glioblastoma: classification and mutational locations. JPMA. The Journal of the Pakistan Medical Association.
2017;
67
(9)
:
1410-1414
.
PubMed Google Scholar -
Liu
A.,
Hou
C.,
Chen
H.,
Zong
X.,
Zong
P.,
Genetics and Epigenetics of Glioblastoma: Applications and Overall Incidence of IDH1 Mutation. Frontiers in Oncology.
2016;
6
(1)
:
16
.
View Article PubMed Google Scholar -
Louis
D.N.,
Perry
A.,
Wesseling
P.,
Brat
D.J.,
Cree
I.A.,
Figarella-Branger
D.,
The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology.
2021;
23
(8)
:
1231-51
.
View Article PubMed Google Scholar -
Siegel
R.L.,
Miller
K.D.,
Fuchs
H.E.,
Jemal
A.,
Cancer Statistics, 2021. CA: a Cancer Journal for Clinicians.
2021;
71
(1)
:
7-33
.
View Article PubMed Google Scholar -
Kruchko
C.,
Ostrom
Q.T.,
Gittleman
H.,
Barnholtz-Sloan
J.S.,
The CBTRUS story: providing accurate population-based statistics on brain and other central nervous system tumors for everyone. Neuro-Oncology.
2018;
20
(3)
:
295-8
.
View Article PubMed Google Scholar -
Duggan
M.A.,
Anderson
W.F.,
Altekruse
S.,
Penberthy
L.,
Sherman
M.E.,
The surveillance, epidemiology, and end results (SEER) program and pathology: toward strengthening the critical relationship. The American Journal of Surgical Pathology.
2016;
40
(12)
:
e94-102
.
View Article PubMed Google Scholar -
Goh
C.H.,
Lu
Y.Y.,
Lau
B.L.,
Wong
J.O.,
Lee
H.K.,
Liew
D.N.,
Brain and spinal tumour. The Medical Journal of Malaysia.
2014;
69
(6)
:
261-267
.
PubMed Google Scholar -
Komori
T.,
The 2016 WHO classification of tumours of the central nervous system: the major points of revision. Neurologia Medico-Chirurgica.
2017;
57
(7)
:
301-11
.
View Article PubMed Google Scholar -
Wesseling
P.,
Capper
D.,
WHO 2016 Classification of gliomas. Neuropathology and Applied Neurobiology.
2018;
44
(2)
:
139-50
.
View Article PubMed Google Scholar -
Ronellenfitsch
M.W.,
Oh
J.E.,
Satomi
K.,
Sumi
K.,
Harter
P.N.,
Steinbach
J.P.,
CASP9 germline mutation in a family with multiple brain tumors. Brain Pathology (Zurich, Switzerland).
2018;
28
(1)
:
94-102
.
View Article PubMed Google Scholar -
Mohamed Yusoff
A.A.,
Zulfakhar
F.N.,
Sul'ain
M.D.,
Idris
Z.,
Abdullah
J.M.,
Association of the IDH1 C.395G > A (R132H) mutation with histological type in malay brain tumors. Asian Pacific Journal of Cancer Prevention.
2016;
17
(12)
:
5195-201
.
PubMed Google Scholar -
Clark
O.,
Yen
K.,
Mellinghoff
I.K.,
Molecular pathways: isocitrate dehydrogenase mutations in cancer. Clinical Cancer Research.
2016;
22
(8)
:
1837-42
.
View Article PubMed Google Scholar -
Liu
Z.,
Che
P.,
Mercado
J.J.,
Hackney
J.R.,
Friedman
G.K.,
Zhang
C.,
Characterization of iPSCs derived from low grade gliomas revealed early regional chromosomal amplifications during gliomagenesis. Journal of Neuro-Oncology.
2019;
141
(2)
:
289-301
.
View Article PubMed Google Scholar -
Agarwal
S.,
Sharma
M.C.,
Jha
P.,
Pathak
P.,
Suri
V.,
Sarkar
C.,
Comparative study of IDH1 mutations in gliomas by immunohistochemistry and DNA sequencing. Neuro-Oncology.
2013;
15
(6)
:
718-26
.
View Article PubMed Google Scholar -
Nejo
T.,
Tanaka
S.,
Ikemura
M.,
Nomura
M.,
Takayanagi
S.,
Shin
M.,
Maffucci syndrome complicated by three different central nervous system tumors sharing an IDH1 R132C mutation: case report. Journal of Neurosurgery.
2018;
131
(6)
:
1829-34
.
View Article PubMed Google Scholar -
Goh
W.C.,
Idris
B.,
Kandasamy
R.,
Shamsuddin
S.,
Jaafar
H.,
Ahmad
F.,
PCR-RFLP method enhance DNA sequencing of IDH1 somatic mutations detection in gliomas. Gulhane Med J.
2020;
61
(4)
:
167-71
.
View Article Google Scholar -
Li
K.,
Ouyang
L.,
He
M.,
Luo
M.,
Cai
W.,
Tu
Y.,
IDH1 R132H mutation regulates glioma chemosensitivity through Nrf2 pathway. Oncotarget.
2017;
8
(17)
:
28865-79
.
View Article PubMed Google Scholar -
Oltvai
Z.N.,
Harley
S.E.,
Koes
D.,
Michel
S.,
Warlick
E.D.,
Nelson
A.C.,
Assessing acquired resistance to IDH1 inhibitor therapy by full-exon IDH1 sequencing and structural modeling. Cold Spring Harbor Molecular Case Studies.
2021;
7
(2)
:
a006007
.
View Article PubMed Google Scholar -
Olafson
L.R.,
Gunawardena
M.,
Nixdorf
S.,
McDonald
K.L.,
Rapkins
R.W.,
The role of TP53 gain-of-function mutation in multifocal glioblastoma. Journal of Neuro-Oncology.
2020;
147
(1)
:
37-47
.
View Article PubMed Google Scholar -
Noor
H.,
Briggs
N.E.,
McDonald
K.L.,
Holst
J.,
Vittorio
O.,
TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers .
2021;
13
(21)
:
5362
.
View Article Google Scholar -
Nordfors
K.,
Haapasalo
J.,
Afyounian
E.,
Tuominen
J.,
Annala
M.,
Häyrynen
S.,
Whole-exome sequencing identifies germline mutation in TP53 and ATRX in a child with genomically aberrant AT/RT and her mother with anaplastic astrocytoma. Cold Spring Harbor Molecular Case Studies.
2018;
4
(2)
:
a002246
.
View Article PubMed Google Scholar -
Ham
S.W.,
Jeon
H.Y.,
Jin
X.,
Kim
E.J.,
Kim
J.K.,
Shin
Y.J.,
TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death and Differentiation.
2019;
26
(3)
:
409-25
.
View Article PubMed Google Scholar -
Kawasoe
T.,
Takeshima
H.,
Yamashita
S.,
Mizuguchi
S.,
Fukushima
T.,
Yokogami
K.,
Detection of p53 mutations in proliferating vascular cells in glioblastoma multiforme. Journal of Neurosurgery.
2015;
122
(2)
:
317-23
.
View Article PubMed Google Scholar -
Ozdogan
S.,
Kafadar
A.,
Yilmaz
S.G.,
Timirci-Kahraman
O.,
Gormus
U.,
Isbir
T.,
Role of caspase-9 gene Ex5+32 G>A (rs1052576) variant in susceptibility to primary brain tumors. Anticancer Research.
2017;
37
(9)
:
4997-5000
.
PubMed Google Scholar -
Kaminska
B.,
Czapski
B.,
Guzik
R.,
Król
S.K.,
Gielniewski
B.,
Consequences of IDH1/2 mutations in gliomas and an assessment of inhibitors targeting mutated IDH proteins. Molecules (Basel, Switzerland).
2019;
24
(5)
:
968
.
View Article PubMed Google Scholar -
Miyata
S.,
Tominaga
K.,
Sakashita
E.,
Urabe
M.,
Onuki
Y.,
Gomi
A.,
Comprehensive Metabolomic Analysis of IDH1 R132H Clinical Glioma Samples Reveals Suppression of β-oxidation Due to Carnitine Deficiency. Scientific Reports.
2019;
9
(1)
:
9787
.
View Article Google Scholar -
Sonoda
Y.,
Kumabe
T.,
Nakamura
T.,
Saito
R.,
Kanamori
M.,
Yamashita
Y.,
Analysis of IDH1 and IDH2 mutations in Japanese glioma patients. Cancer Science.
2009;
100
(10)
:
1996-8
.
View Article PubMed Google Scholar -
Cerami
E.,
Gao
J.,
Dogrusoz
U.,
Gross
B.E.,
Sumer
S.O.,
Aksoy
B.A.,
The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery.
2012;
2
(5)
:
401-4
.
View Article PubMed Google Scholar -
Molenaar
R.J.,
Maciejewski
J.P.,
Wilmink
J.W.,
Noorden
C.J. Van,
Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene.
2018;
37
(15)
:
1949-60
.
View Article PubMed Google Scholar -
Park
J.W.,
Turcan
S.,
Epigenetic reprogramming for targeting IDH-mutant malignant gliomas. Cancers (Basel).
2019;
11
(10)
:
1616
.
View Article PubMed Google Scholar -
Núñez
F.J.,
Mendez
F.M.,
Kadiyala
P.,
Alghamri
M.S.,
Savelieff
M.G.,
Koschmann
C.,
IDH1R132H acts as a tumor suppressor in glioma via epigenetic upregulation of the DNA damage response. Sci Transl Med.
2018;
11
(479)
:
eaaq1427
.
View Article PubMed Google Scholar -
Park
J.W.,
Turcan
S.,
Epigenetic reprogramming for targeting IDH-mutant malignant gliomas. Cancers (Basel).
2019;
11
(10)
:
1616
.
View Article PubMed Google Scholar -
Zhao
S.,
Lin
Y.,
Xu
W.,
Jiang
W.,
Zhai
Z.,
Wang
P.,
Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science (1979).
2009;
324
(5924)
:
261-265
.
View Article PubMed Google Scholar -
Turcan
S.,
Rohle
D.,
Goenka
A.,
Walsh
L.A.,
Fang
F.,
Yilmaz
E.,
IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature.
2012;
483
(7390)
:
479-83
.
View Article PubMed Google Scholar -
England
B.,
Huang
T.,
Karsy
M.,
Current understanding of the role and targeting of tumor suppressor p53 in glioblastoma multiforme. Tumour Biology.
2013;
34
(4)
:
2063-74
.
View Article PubMed Google Scholar -
Tang
C.,
Chua
C.L.,
Ang
B.T.,
Insights into the cancer stem cell model of glioma tumorigenesis. Annals of the Academy of Medicine, Singapore.
2007;
36
(5)
:
352-7
.
PubMed Google Scholar -
Parada
L.F.,
Dirks
P.B.,
Wechsler-Reya
R.J.,
Brain tumor stem cells remain in play. Journal of Clinical Oncology.
2017;
35
(21)
:
2428-31
.
View Article PubMed Google Scholar -
Gulaia
V.,
Kumeiko
V.,
Shved
N.,
Cicinskas
E.,
Rybtsov
S.,
Ruzov
A.,
Molecular mechanisms governing the stem cell's fate in brain cancer: factors of stemness and quiescence. Frontiers in Cellular Neuroscience.
2018;
12
:
388
.
View Article PubMed Google Scholar -
Llaguno
S.A.,
Chen
J.,
Kwon
C.H.,
Jackson
E.L.,
Li
Y.,
Burns
D.K.,
Malignant Astrocytomas Originate from Neural Stem/Progenitor Cells in a Somatic Tumor Suppressor Mouse Model. Cancer Cell.
2009;
15
(1)
:
45-56
.
View Article Google Scholar -
Fantin
V.,
Dang
L.,
White
D.,
Gross
S.,
Bittinger
M.,
Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Yearbook of Neurology and Neurosurgery.
2010;
2010
:
111-2
.
View Article Google Scholar -
Shi
J.,
Sun
B.,
Shi
W.,
Zuo
H.,
Cui
D.,
Ni
L.,
Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biology.
2015;
36
(2)
:
655-62
.
View Article PubMed Google Scholar -
Sancho-Martinez
I.,
Nivet
E.,
Xia
Y.,
Establishment of human iPSC-based models for the study and targeting of glioma initiating cells. Nat Commun.
2016;
7
:
10743
.
View Article Google Scholar -
van Pel
D.M.,
Harada
K.,
Song
D.,
Naus
C.C.,
Sin
W.C.,
Modelling glioma invasion using 3D bioprinting and scaffold-free 3D culture. Journal of Cell Communication and Signaling.
2018;
12
(4)
:
723-30
.
View Article PubMed Google Scholar -
Lancaster
M.,
Knoblich
J.,
Generation of cerebral organoids from human pluripotent stem cells . Nature Protocols.
2017;
9
(10)
:
2329-40
.
View Article PubMed Google Scholar -
Linkous
A.,
Balamatsias
D.,
Snuderl
M.,
Edwards
L.,
Miyaguchi
K.,
Milner
T.,
Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Reports.
2019;
26
(12)
.
View Article PubMed Google Scholar -
Ogawa
J.,
Pao
G.M.,
Shokhirev
M.N.,
Verma
I.M.,
Glioblastoma Model Using Human Cerebral Organoids. Cell Reports.
2018;
23
(4)
:
1220-9
.
View Article PubMed Google Scholar -
Zhang
L.,
Liu
F.,
Weygant
N.,
Zhang
J.,
Hu
P.,
Qin
Z.,
A novel integrated system using patient-derived glioma cerebral organoids and xenografts for disease modeling and drug screening. Cancer Letters.
2021;
500
:
87-97
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 9 No 10 (2022)
Page No.: 5375-5383
Published on: 2022-10-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 4931 times
- PDF downloaded - 1405 times
- XML downloaded - 0 times
