

Copyrights: Huong Thu Duong, Phuong Minh Pham, Nam Huynh Bao Tran, Phuong Thanh Huynh, Hung Trong Hoang, Thinh Huy Quoc Dang, Tu Anh Thai, Chi Thi Kim Nguyen, Nam Cong Nhat Huynh, 2023. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Introduction: Formalin-fixed paraffin-embedded (FFPE) tissue provides a valuable source of information for pathological studies and oral cancer pathology. However, FFPE tissue immobilization and storage often cause the partial degradation of nucleic acids, resulting in mRNA sequencing libraries that may not be of sufficient quantity and quality for gene expression analysis. We optimized the RNA extraction and library preparation process to increase the amount of useful data obtained with low-quality RNA from FFPE oral cancer tissue samples.
Methods: This study used 20 samples stored for 1 - 2 years. After RNA extraction from FFPE samples, we compared two methods for library preparation, rRNA depletion, and exome capture, to make recommendations for metrics such as RNA input and output concentrations and generated full RNA sequencing data for downstream bioinformatics analysis.
Results: The quantity of RNA extracted from six 8-mm-thick slices of FFPE tissue was sufficient for library preparation (130 ng/µL); sample quality did not differ significantly with storage time. Additionally, the RNA samples had an average DV200 index of 30% - 50%. Exome capture outperformed rRNA depletion for library preparation in library output concentration (p < 0.001) and RNA sequencing data generated for bioinformatics analysis.
Conclusion: RNA can be extracted from FFPE samples for sequencing, provided they have been handled and stored appropriately. Exome capture is the best method for preparing libraries for RNA sequencing from low-quality tissue samples such as FFPE.
Introduction
Oral squamous cell carcinoma (OSCC) is the most common head and neck malignancy, with over 400,000 new cases diagnosed annually1. OSCC development and progression are complicated by the interaction and influence of many genes and other signaling pathways2, 3. There remains a lack of biomarkers and targeted therapies for OSCC. Therefore, searching for targeted therapies for OSCC becomes especially important.
Next-generation RNA sequencing (RNA-seq) has been widely used in cancer research4. Previous molecular profiles of common malignancies in The Cancer Genome Atlas have been generated from fresh tumor samples, which yielded high-quality RNA for qualitative analyses5. However, for any cancer, only some cases have fresh tissues available for unbiased RNA analysis, of which few have long-term treatment data available6. Therefore, researchers have used formalin-fixed paraffin-embedded (FFPE) tissue samples for RNA sequencing, particularly since FFPE tissue biorepositories are maintained at all oncology hospitals and can be linked to detailed patient clinical data to delineate control and trial groups. In addition, FFPE tissue samples are usually stored for at least 10 years, enabling the study of long-term treatment outcomes.
Despite its many advantages, FFPE tissue sample processing and storage can result in highly degraded RNA, limiting gene detection and possibly generating spurious gene sequences. Therefore, researchers have endeavored to improve techniques and procedures for analyzing RNA from FFPE samples7, 8. In this study, we standardize the RNA sequencing process for FFPE tissue samples and identify several factors related to tissue selection, RNA isolation, library selection, and data analysis to ensure the RNAseq data obtained from the FFPE samples is valid. We aimed to compare two commonly used RNA library preparation protocols using low-quality RNA from FFPE oral cancer tissue samples and to recommend RNA input metrics to ensure adequate RNA-seq data for downstream bioinformatics analysis.
Methods
Clinical samples
The study used 20 retrospective FFPE tissue samples from patients with OSCC who were examined and treated at Ho Chi Minh City Oncology Hospital. Among the studied samples, 13 had been stored for one year, and seven had been stored for two years.
For each FFPE sample, one 3–4 µm thick slice was cut for hematoxylin and eosin (H&E) staining, and 4–6 8 µm thick slices were cut for RNA extraction. A pathologist marked the cancer tissue on the H&E slide to locate the corresponding tumor on the 4–6 tissue slices used for RNA extraction. These tumor sites were scraped with a sterile blade, placed in a 1.5 mL Eppendorf tube, and stored at −20°C until needed for RNA extraction.
RNA isolation
RNA was extracted from the FFPE tissue samples using the PureLink FFPE RNA Isolation Kit (K156002; Invitrogen). The extraction procedure comprised the following steps: thermal decomposition of paraffin for 10 minutes, sample lysis with proteinase K for 10 minutes, sample washing for five minutes, and RNA dissolution for two minutes9.
The extracted RNA was evaluated for quantity by measuring its concentration on a QuantiFluor® RNA System (Promega) and for quality by determining its DV200 index10. The DV200 index is the percentage of RNA with a length >200 nucleotides. FFPE samples with a DV200 of >70% are considered high-quality, those with a DV200 of 50%–70% are considered medium-quality, those with a DV200 of 30%–50% are considered low-quality, and those with a DV200 <30% are considered heavily degraded and are excluded from RNA-seq11. Finally, the extracted RNA was stored at −80°C until required for library preparation.
Among the 20 studied samples, eight were used to optimize the number of FFPE slices required for RNA sequencing (four or six 8 µm slices) and determine whether or not to remount the FFPE wax block (remounting makes FFPE tissue slicing easier and more accurate for immunohistochemical staining). The extraction results were evaluated and compared based on the concentration and DV200 index before selecting the optimal method for RNA extraction and applying it to the subsequent samples.
Library preparation and sequencing
This study evaluated two commonly used RNA library preparation protocols for FFPE samples: rRNA depletion and exome capture.
The rRNA depletion method was applied to 10 FFPE samples. The rRNAs (cytoplasmic 5S, 5.8S, 18S, 28S, ITS, human ETS, and 12S and 16S mitochondria rRNA) were removed from the total extracted RNA with the NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat; New England Biolabs), and the rRNA-depleted RNA was used for library preparation with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs) according to the manufacturer's instructions.
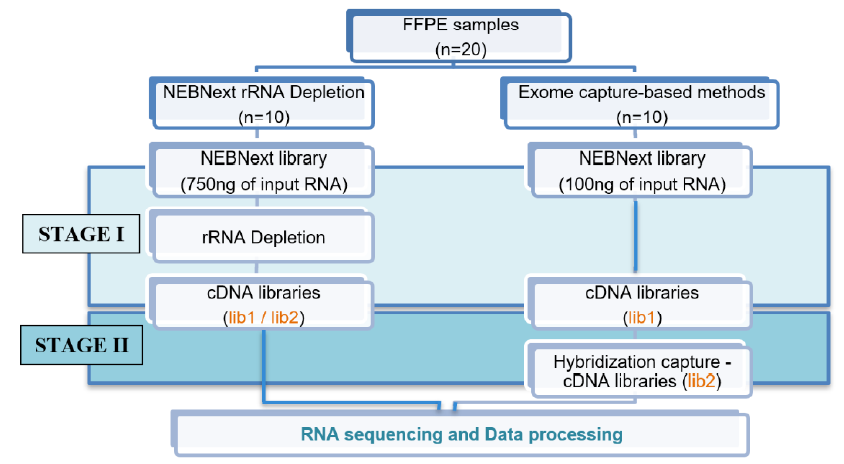
The Exome capture method was applied to the remaining 10 FFPE samples. This method comprised two stages: the first prepares a cDNA library from the extracted RNA with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (omitting the rRNA removal step; New England Biolabs), and the second performs target enrichment using a hybridization procedure with the xGen NGS Hybridization Capture Kit (Integrated DNA Technologies; Figure 1).

The input RNA concentration was 750 ng for rRNA depletion and 100 ng for exome capture. The cDNA levels obtained in the library preparation were measured using a QuantiFluor® DNA System. Here, lib-1 is the cDNA concentration of stage I, and lib-2 is the cDNA concentration of stage II. Because the NEBNext rRNA depletion method only performed stage I, lib-1 and lib-2 were the same for this method. Finally, the libraries were sequenced on an Illumina MiniSeq Sequencing System with a 100-cycle protocol and single-end reads, generating approximately 40 million reads per sample. The sequencing data files were converted to the FASTQ format and then processed in Python (v3.7) using command packages, FastQC (v.0.1.1.8), TrimGalore (v.0.6.4), and Kallisto (v.0.46.0) with a human transcriptome index (Homo sapiens GRCh38).
Statistics analysis
Data are presented as the mean ± standard deviation. Data normality was assessed using the Shapiro–Wilk test. The cDNA concentrations were compared between the two library preparation methods using the nonparametric Mann–Whitney U test. The mRNA concentrations and DV200 indexes were compared by storage year, number of slices, and FFPE remounted procedure using the parametric t-test. A p < 0.05 was considered statistically significant.


| Sample | % rRNA | % mRNA | Insert Size | % Dups | % Aligned | M Aligned |
|---|---|---|---|---|---|---|
| 32_1 | 0.0% | 93.6% | 130 bp | 60.9% | 95.7% | 15.0 |
| 33_27 | 0.7% | 89.4% | 130 bp | 80.1% | 92.3% | 1.3 |
| 34_1 | 0.1% | 94.1% | 155 bp | 53.8% | 93.9% | 18.2 |
| 143_1 | 0.9% | 91.8% | 133 bp | 80.9% | 92.8% | 3.2 |
Results
Sample storage time
The study included 20 samples stored for either one or two years. Group 1 comprised 13 samples stored for one year, and Group 2 comprised seven samples stored for two years. RNA extraction results showed that all samples met RNA quality and quantity for inclusion in the library preparation process, with a concentration higher than 130 ng/µL and a DV200 index higher than 30%. Groups 1 and 2 did not differ significantly (Figure 2 A, B).
Number of slices and FFPE remount procedure
Eight samples were divided into four groups to test the quality and concentration of the extracted RNA. Groups 1 and 2 each comprised two RNA samples extracted from four or six 8 µm thick slices, respectively, cut from blocks that had not been remounted. Groups 3 and 4 each comprised two RNA samples extracted from four or six 8 µm thick slices, respectively, that had been cut from remounted blocks. RNA was extracted from all samples with the PureLink™ FFPE RNA Isolation Kit, providing concentrations >130 ng/µL and DV200 indexes ≥34% for all samples, with no apparent differences in RNA quantity and quality among the four groups (Figure 2 C, D, E, F). This finding shows that the number of slices and the FFPE remounting procedure did not affect RNA extraction. Therefore, we chose to perform FFPE block remounting before slicing and used six 8 µm thick slices for all subsequent FFPE samples.
Library preparation and sequencing
The amount of RNA obtained from the extraction procedure was higher than the amount of input RNA required for both library preparation methods. We used 750 ng of input RNA for the rRNA depletion method and 100 ng of input RNA for the exome capture method; this input RNA amount was adjusted as recommended by the standard procedure of each kit.
After the rRNA depletion procedure, the cDNA concentration was quite low for the 10 samples, ranging from 0.35 to 1.53 ng/µL, so the amount of cDNA obtained after the library preparation procedure was between 7.0 and 30.6 ng. As a routine procedure, we conduct 14 cycles of PCR amplification for the NEBNext rRNA depletion library.
The exome capture-based library preparation method comprised two stages: stage I prepared the cDNA library from the extracted RNA with the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (omitting the rRNA removal step), and stage II performed target enrichment using a hybridization process with the xGen Hybridization Capture Kit. The cDNA concentration was determined after stage I (lib-1), and this stage was repeated from 1 to >2 times for each sample to achieve a minimum cDNA amount required for the hybridization procedure (250 ng). The stage I cDNA concentrations of the 10 samples ranged from 3.11 to 65.8 ng/µL, with eight reaching >250 ng after one preparation, two after two preparations, and one after three preparations and a three-cycle PCR amplification.
The stage I cDNA concentrations (lib1) were significantly higher with the exome capture-based method than with the NEBNext rRNA depletion method (p < 0.001), possibly because the total RNA was handled less in the exome-capture method (reverse transcription only) than in the rRNA depletion method (rRNA depletion and reverse transcription), providing less opportunity for it to degrade.
After conducting stage I of the exome capture-based method, we continued with stage II, which involved target enrichment via hybridization with the xGen Hybridization Capture Kit with 250 ng of input cDNA for each sample. After the hybridization step, the cDNA concentration (lib-2) ranged from 9.50 to 13.25 ng/µL across samples, a high concentration that should provide good sequencing results. The cDNA concentration differed significantly between the rRNA depletion and exome capture-based methods (p < 0.001), suggesting the exome capture-based method performs better in RNA library preparation (Figure 3).
After RNA-seq, the 10 samples prepared using the exome capture-based method had better results than those prepared using the NEBNext rRNA depletion method. The percentage of aligned reads on the human genome was about ≥90% in each sample, with 89%–94% of reads mapping to mRNA regions and <1% mapping to rRNA regions. The mRNA regions mainly comprise coding sequence regions (accounting for 79% – 87%; Table 1).
The RNA-seq results of the samples prepared with the NEBNext rRNA depletion method showed a rather short fragment size distribution due to the adapter in the sample. The insert size fluctuated between 100 bp to <50 bp. In addition, only about 10% – 30% of the reads mapped to the human genome, with the rest unmapped. The unmapped reads were marked too short because less than two-thirds of their length was aligned to the reference sequence. The number of reads mapped to mRNA regions was quite low, <2000. Principal coordinate analysis (PCA) is unreliable when so few reads are used.
Discussion
Clinical biological samples are often stored as FFPE blocks, representing an invaluable resource for biomedical research. Such FFPE blocks allow long-term clinical sample storage, preserving tissue morphology and nucleic acid information12, 13. However, gene expression analysis can be more challenging with RNA extracted from FFPE samples than from other sources due to RNA degradation. Sample preservation with formalin hinders several molecular applications. In addition, FFPE processing and tissue storage have been shown to affect RNA quality, limiting the quantification of gene expression by technologies such as RNA-seq14. Another challenge with using FFPE samples is that standard RNA extraction workflows often provide poor RNA quality and quantity15. Etiologically, formaldehyde induces the formation of adducts and crosslinks and interferes with the analysis of RNA itself16. In addition, the RNA isolated from FFPE samples is typically fragmented, and its utility might also be hampered by contaminants and inhibitors17. Therefore, it is vital to establish a robust and reproducible method for extracting RNA of sufficient quality and quantity for downstream analyses. Our study provides a guide for further studies using FFPE samples for RNA-seq. By following our recommendations, sequencing samples with RNA and library inputs higher than our suggested values will result in a higher success rate for RNA-seq and reduce unnecessary sequencing costs.
Our result shows that RNA quantity and quality of RNA did not significantly differ between samples stored for one or two years (p > 0.05). Choi et al. also reported similar results for RNA quality from FFPE samples stored for up to 10 years, which could be used for RNA-seq. However, FFPE samples should be checked carefully and immobilized within six hours after surgery18. Yi et al. noted that specimens that were stored for longer were more degraded and provided lower RNA yields than others, but there was no significant difference in RNA purity19.
In our study, the amount of RNA extracted from FFPE samples was higher than 750 ng, ensuring sufficient input for library preparation, and did not differ between remounted and non-remounted FFPE blocks and the amount of tissue (p > 0.05). However, Jarzab et al. noted that isolation efficacy was lower when three sections were used (178 ng/µL) compared to 5–8 sections (279 and 301 ng/µL, respectively)20. Therefore, we chose to remount the FFPE block and cut six 8 µm thick slices for all FFPE samples in our study to ensure more favorable slice cutting for histochemical staining and RNA extraction, assuring adequate RNA amounts.
In our study, we used the minimum amount of RNA for the NEBNext rRNA depletion library method (750 ng) and the exome capture-based library method (100 ng). The exome capture-based method is preferable for degraded FFPE samples with low RNA extraction quality and quantity due to its smaller input requirement (100 ng). Indeed, standard RNA-seq protocols usually require a relatively large amount of input RNA, making them difficult to apply to scarce and degraded RNA from fixed clinical samples. Wang et al. investigated the percentage of mapped reads, finding the percentages of known splice alignments, partly known splice alignments, and novel splice alignments to be relatively comparable across all RNA input levels21. Therefore, the exome capture-based library preparation method that requires lower input is more commonly used for FFPE samples.
The cDNA concentration of the libraries prepared with the NEBNext rRNA depletion method ranged from 0.35 to 1.53 ng/µL across samples. In contrast, the cDNA concentrations of libraries prepared with the exome capture-based method ranged from 9.5 to 13.25 ng/µL across samples, significantly higher than those with the NEBNext rRNA depletion method. The RNA-seq results showed that for the low-quality RNA samples, such as those in our study, the NEBNext rRNA depletion library method provided relatively few usable reads for analysis, leading to unstable PCA results, due to short fragment sizes and poorer quality. Therefore, this result is also similar to that of Lin et al.8. The mRNA capture method used during rRNA knockdown (cytoplasmic 5S, 5.8S, 18S, 28S, ITS, human ETS, and 12S and 16S mitochondria) has a loose junction that can also remove mRNAs, reducing the amount of mRNA available for reverse transcription into cDNA and PCR amplification, making the amount of cDNA in the library quite low and of uncertain quality.
With the exome capture-based method, all RNAs in the extracted sample are reverse transcribed into cDNA, which is more stable than the original RNA, and amplified with 14 PCR cycles, facilitating hybridization with the xGen Hybridization Capture Kit. This hybridization does not use conventional wash beads like the NEBNext rRNA depletion method. Instead, streptavidin beads form strong bonds with streptavidin-5'-biotinylated primers on xGen Lockdown Probes22, facilitating the purification process and improving the library preparation. However, the exome capture-based library preparation method has some disadvantages, such as the need to wash the streptavidin beads at 65°C to ensure complete disassembly of the streptavidin-5'-biotinylated oligo junction. This method also takes a long time because it requires two stages (cDNA amplification and hybridization). According to some studies, the exome capture-based hybridization method helps to significantly increase the amount of exon data due to cDNA capture. However, the accuracy of quantified gene expression decreased8, 14, 23.
A limitation of our study was its sample size. We plan to confirm our findings in a larger cohort to ensure that there is sufficient power to detect statistical significance and test samples from different disease models.
Conclusions
In summary, our results show that mRNA sequencing of FFPE samples with storage times of 1–2 years has a high success rate. The NEBNext rRNA depletion and exome capture-based library preparation methods can both be used for good-quality FFPE samples. However, the exome capture-based library preparation method is optimal for samples with low RNA quantity and quality or appreciable degradation.
Abbreviations
xxx
Acknowledgments
We thank Ho Chi Minh City Oncology Hospital, Gene Solutions, Medical Genetics Institute, and Center for Molecular Biomedicine, The University of Medicine and Pharmacy at Ho Chi Minh City, Vietnam for supporting this study.
Author’s contributions
All authors read and approved the final manuscript.
Funding
This study was granted by the Department of Science and Technology (DOST), Ho Chi Minh City, Vietnam (No. 07/2022/HĐ-QKHCN).
Availability of data and materials
Data and Materials are used and analyzed during the study being available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The study was approved by the University of Medicine and Pharmacy at Ho Chi Minh City (No. 441/2021).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Chiou
S.H.,
Yu
C.C.,
Huang
C.Y.,
Lin
S.C.,
Liu
C.J.,
Tsai
T.H.,
Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clinical Cancer Research.
2008;
14
(13)
:
4085-95
.
View Article PubMed Google Scholar -
Sung
H.,
Ferlay
J.,
Siegel
R.L.,
Laversanne
M.,
Soerjomataram
I.,
Jemal
A.,
Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: a Cancer Journal for Clinicians.
2021;
71
(3)
:
209-49
.
View Article PubMed Google Scholar -
Zhang
Y.Y.,
Mao
M.H.,
Han
Z.X.,
Identification of a Gene Prognostic Signature for Oral Squamous Cell Carcinoma by RNA Sequencing and Bioinformatics. BioMed Research International.
2021;
2021
:
6657767
.
View Article PubMed Google Scholar -
Pillai
J.,
Chincholkar
T.,
Dixit
R.,
Pandey
M.,
A systematic review of proteomic biomarkers in oral squamous cell cancer. World Journal of Surgical Oncology.
2021;
19
(1)
:
315
.
View Article PubMed Google Scholar -
Cancer Genome Atlas
N.,
Cancer Genome Atlas Network
Comprehensive molecular portraits of human breast tumours. Nature.
2012;
490
(7418)
:
61-70
.
View Article PubMed Google Scholar -
Pennock
N.D.,
Jindal
S.,
Horton
W.,
Sun
D.,
Narasimhan
J.,
Carbone
L.,
RNA-seq from archival FFPE breast cancer samples: molecular pathway fidelity and novel discovery. BMC Medical Genomics.
2019;
12
(1)
:
195
.
View Article PubMed Google Scholar -
Li
J.,
Fu
C.,
Speed
T.P.,
Wang
W.,
Symmans
W.F.,
Accurate RNA Sequencing From Formalin-Fixed Cancer Tissue To Represent High-Quality Transcriptome From Frozen Tissue. JCO Precision Oncology.
2018;
2018
.
View Article Google Scholar -
Lin
X.,
Qiu
L.,
Song
X.,
Hou
J.,
Chen
W.,
Zhao
J.,
A comparative analysis of RNA sequencing methods with ribosome RNA depletion for degraded and low-input total RNA from formalin-fixed and paraffin-embedded samples. BMC Genomics.
2019;
20
(1)
:
831
.
View Article PubMed Google Scholar -
Wehmas
L.C.,
Wood
C.E.,
Gagne
R.,
Williams
A.,
Yauk
C.,
Gosink
M.M.,
Demodifying RNA for Transcriptomic Analyses of Archival Formalin-Fixed Paraffin-Embedded Samples. Toxicological Sciences.
2018;
162
(2)
:
535-47
.
View Article PubMed Google Scholar -
Matsubara
T.,
Soh
J.,
Morita
M.,
Uwabo
T.,
Tomida
S.,
Fujiwara
T.,
DV200 Index for Assessing RNA Integrity in Next-Generation Sequencing. BioMed Research International.
2020;
2020
:
9349132
.
View Article PubMed Google Scholar -
Wang
D.,
Rolfe
P.A.,
Foernzler
D.,
O'Rourke
D.,
Zhao
S.,
Scheuenpflug
J.,
Comparison of Two Illumina Whole Transcriptome RNA Sequencing Library Preparation Methods Using Human Cancer FFPE Specimens. Technology in Cancer Research {&}amp; Treatment.
2022;
21
:
15330338221076304
.
View Article PubMed Google Scholar -
Marczyk
M.,
Fu
C.,
Lau
R.,
Du
L.,
Trevarton
A.J.,
Sinn
B.V.,
The impact of RNA extraction method on accurate RNA sequencing from formalin-fixed paraffin-embedded tissues. BMC Cancer.
2019;
19
(1)
:
1189
.
View Article PubMed Google Scholar -
Amini
P.,
Ettlin
J.,
Opitz
L.,
Clementi
E.,
Malbon
A.,
Markkanen
E.,
An optimised protocol for isolation of RNA from small sections of laser-capture microdissected FFPE tissue amenable for next-generation sequencing. BMC Molecular Biology.
2017;
18
(1)
:
22
.
View Article PubMed Google Scholar -
Cieslik
M.,
Chugh
R.,
Wu
Y.M.,
Wu
M.,
Brennan
C.,
Lonigro
R.,
The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Research.
2015;
25
(9)
:
1372-81
.
View Article PubMed Google Scholar -
Patel
P.G.,
Selvarajah
S.,
Guérard
K.P.,
Bartlett
J.M.,
Lapointe
J.,
Berman
D.M.,
Reliability and performance of commercial RNA and DNA extraction kits for FFPE tissue cores. PLoS One.
2017;
12
(6)
:
e0179732
.
View Article PubMed Google Scholar -
Groelz
D.,
Sobin
L.,
Branton
P.,
Compton
C.,
Wyrich
R.,
Rainen
L.,
Non-formalin fixative versus formalin-fixed tissue: a comparison of histology and RNA quality. Experimental and Molecular Pathology.
2013;
94
(1)
:
188-94
.
View Article PubMed Google Scholar -
Okello
J.B.,
Zurek
J.,
Devault
A.M.,
Kuch
M.,
Okwi
A.L.,
Sewankambo
N.K.,
Comparison of methods in the recovery of nucleic acids from archival formalin-fixed paraffin-embedded autopsy tissues. Analytical Biochemistry.
2010;
400
(1)
:
110-7
.
View Article PubMed Google Scholar -
Choi
Y.,
Kim
A.,
Kim
J.,
Lee
J.,
Lee
S.Y.,
Kim
C.,
Optimization of RNA Extraction from Formalin-Fixed Paraffin-Embedded Blocks for Targeted Next-Generation Sequencing. Journal of Breast Cancer.
2017;
20
(4)
:
393-9
.
View Article PubMed Google Scholar -
Yi
Q.Q.,
Yang
R.,
Shi
J.F.,
Zeng
N.Y.,
Liang
D.Y.,
Sha
S.,
Effect of preservation time of formalin-fixed paraffin-embedded tissues on extractable DNA and RNA quantity. The Journal of International Medical Research.
2020;
48
(6)
:
300060520931259
.
View Article PubMed Google Scholar -
Jarzab
M.,
Rózanowski
P.,
Kowalska
M.,
Zebracka
J.,
Rudnicka
L.,
Stobiecka
E.,
Optimization of the method of RNA isolation from paraffin blocks to assess gene expression in breast cancer. Polish Journal of Pathology.
2008;
59
(2)
:
85-91
.
PubMed Google Scholar -
Wang
L.,
Felts
S.J.,
Van Keulen
V.P.,
Pease
L.R.,
Zhang
Y.,
Exploring the effect of library preparation on RNA sequencing experiments. Genomics.
2019;
111
(6)
:
1752-9
.
View Article PubMed Google Scholar -
Steiert
T.A.,
Fu\ss
J.,
Juzenas
S.,
Wittig
M.,
Hoeppner
M.P.,
Vollstedt
M.,
High-throughput method for the hybridisation-based targeted enrichment of long genomic fragments for PacBio third-generation sequencing. NAR Genomics and Bioinformatics.
2022;
4
(3)
.
View Article PubMed Google Scholar -
Herbert
Z.T.,
Kershner
J.P.,
Butty
V.L.,
Thimmapuram
J.,
Choudhari
S.,
Alekseyev
Y.O.,
Cross-site comparison of ribosomal depletion kits for Illumina RNAseq library construction. BMC Genomics.
2018;
19
(1)
:
199
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 10 No 10 (2023)
Page No.: 5987-5994
Published on: 2023-10-31
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
- HTML viewed - 4512 times
- PDF downloaded - 1341 times
- XML downloaded - 132 times
