

The proliferation of MDA-MB-231 cells is repressed by mesenchymal stem cell-mediated macrophage activation conditioned medium through the inhibition of AKT1 and YKL-39 genes
-
Nur Ramziahrazanah Jumat

-
Muhammad Amir Yunus
-
Badrul Hisham Yahaya
-
Mohd Yusmaidie Aziz
-
Mohd Salleh Rofiee
-
Rafeezul Mohamed
Copyrights: Nur Ramziahrazanah Jumat, Muhammad Amir Yunus, Badrul Hisham Yahaya, Mohd Yusmaidie Aziz, Mohd Salleh Rofiee, Rafeezul Mohamed, 2024. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Background: Triple-negative breast cancer (TNBC) is characterized by a substantial presence of tumor-associated macrophages (TAMs) exhibiting an M2-like phenotype, which plays a crucial role in promoting tumor cell stemness and invasiveness. Mesenchymal stem cells (MSCs) have the ability to induce the transformation of naive macrophages (M0) into M1-like macrophages. This study delves into the interplay between MSCs and macrophages within the context of breast cancer (BC) progression using a TNBC cell line, as reprogramming of TAMs into M1-like macrophages may emerge as a promising therapeutic strategy for BC.
Methods: THP-1 cells were induced into M0 macrophages and co-cultured with UC-MSCs, subsequently analyzing CM for M1- and M2-type macrophage-related cytokines. Total RNA from co-cultured cells was used to assess IRF-4 and IRF- 5 mRNA gene expression via qRT-PCR. MDA-MB-231 cells were exposed to CM and co-cultured cells to evaluate cell viability through MTT assay over 24, 48, and 72 hours, with qRT-PCR used to examine breast cancer-related gene expression.
Results: The results indicate that co-culturing M0 macrophages with MSCs promotes M1-like macrophages, as evidenced by upregulated IRF- 5 and suppressed M2 macrophage-related genes. Treatment with CM from M0/MSCs co-culture significantly inhibits MDA-MB-231 cell proliferation at 72 hours, accompanied by reduced TNF-alpha levels. Notably, CM treatment downregulates AKT1 and YKL-39 genes in MDA-MB-231 cells, suggesting potential anti-cancer effects. Direct co-culture with M0/MSCs, however, shows no significant impact on TNBC cell growth.
Conclusion: This study highlights MSCs' ability to induce M0 macrophages to a M1-like phenotype and suggests that CM from M0/MSCs co-culture may contain anti-cancer factors targeting AKT1 and YKL-39 genes, underscoring the potential of MSC-mediated macrophage activation as a strategy to enhance BC treatment, especially in the context of TNBC.
Introduction
Breast cancer (BC) is the primary factor responsible for fatalities in women, with approximately 2.3 million newly diagnosed cases and 685,000 reported deaths worldwide in 20201. The mortality rate of BC is significant, contributing to 16% or 1 in every 6 cancer-related deaths in females. The main reason is primarily attributed to the spread of the disease to other parts of the body and the challenge of overcoming systemic treatments1. In the beginning, research into BC metastasis and treatment resistance primarily focused on tumor cells alone. Nevertheless, recent years have witnessed thorough investigations into how the tumor microenvironment (TME) contributes to advancing distant metastasis and resistance to therapy2. This shift in focus has revealed the significance of non-malignant cells and TME components, such as immune cells and the extracellular matrix, in BC progression3. As a result, diverse approaches have undergone investigation to target these non-malignant cells and TME constituents, aiming to disrupt the tumor-promoting environment and enhance treatment outcomes3. By understanding the complex interplay between tumor cells and their microenvironment, novel therapeutic approaches can be developed to combat BC metastasis and overcome treatment resistance, offering new hope for patients worldwide.
In normal physiological conditions, tissue-resident macrophages serve as innate immune cells that contribute to phagocytic capability, sustaining tissue homeostasis, and defending against pathogens4. However, in the context of BC, tumor-associated macrophages (TAMs) heavily infiltrate the TME, comprising over 50% of the cell population within the TME4. The TME in BC consists of various components, including adipocytes, fibroblasts, and various categories of leukocytes such as dendritic cells, lymphocytes, and neutrophils5. The recruitment of circulating monocytes and resident macrophages sustains the population of TAMs in BC6. Upon recruitment, monocytes differentiate into naïve macrophages (M0) under the stimulation of monocyte colony-stimulating factor6. Macrophages can polarize into two main phenotypes: M1-like macrophages, also known as classically activated macrophages, and M2-like macrophages, also referred to as alternatively activated macrophages7. M1-like macrophages are stimulated by T helper 1-type cytokines such as interferon gamma (IFNγ) or tumor necrosis factor-alpha (TNF-α) and exhibit anti-tumor responses. They secrete pro-inflammatory cytokines like TNF-α and IL-2, along with reactive nitrogen and oxygen intermediates7. Conversely, M2-like macrophages are induced by T helper 2-type cytokines such as IL-4, IL-10, and IL-13 and demonstrate pro-tumor responses7. Under the influence of specific stimuli, various subtypes of M2a, M2b, M2c, and M2d can be induced from M2-like macrophages8. In the TME, cancer cells release cytokines that attract TAMs, which closely resemble M2 macrophages9. TAMs, in turn, impede the invasion and role of anti-tumor CD8+ T cells (CTLs), promote tumor angiogenesis, and stimulate tumor cell proliferation and metastasis9. The most effective strategy in cancer prevention research has recently shifted from macrophage reduction to TAM re-education10. Due to their great ability and flexibility to adjust to external signals, macrophages can have their biological functions taken over and altered in cancer10. It is crucial to evaluate TAMs holistically, considering their ontogeny, TME-mediated education, phenotypic diversity, placement within the TME, and tumor-modulating roles10. Consequently, targeting the recruitment of TAMs or reconfiguring them into a phenotype competent to eliminate tumor cells has emerged as a potential therapeutic approach in BC.
Mesenchymal stem cells (MSCs) belong to the mesoderm lineage and encompass various multipotent cells, including adipocytes, chondrocytes, fibroblasts, osteoblasts, and smooth muscle cells, among others11. MSCs are characterized by the expression of CD73, CD90, and CD105, while lacking hematopoietic lineage markers such as CD14, CD20, CD34, CD45, and HLA-DR11. Extensive evidence suggests that MSCs foster the advancement of cancer via numerous avenues, encompassing the stimulation of blood vessel formation, infiltration, proliferation, viability, the creation of carcinoma-associated fibroblasts, and the hindrance of natural and acquired anti-tumor reactions12. However, MSCs may even inhibit tumor growth. MSC-CM has been shown to limit breast cancer cell proliferation and make cancer cells more sensitive to radiation by suppressing the STAT3 signaling pathway13.
The relationship that exists between tissue-draining macrophages and allogeneic MSCs has been explored, in which the macrophages were transformed into regulatory macrophages that inhibited T cell responses in vivo14. Recent research indicates that the interaction between MSCs and macrophages can promote macrophage polarization to the M2-like phenotype when triggered in the M1 activation stage15. MSC-derived soluble factors can convert monocytes or M1 macrophages into M2 cells, with PGE2 playing a significant role16. Additionally, a study by Vasandan et al. revealed that MSCs induce respiratory burst and nitric oxide-dependent killing mechanisms in macrophages17. MSCs altered naïve macrophages towards a pro-inflammatory M1 polarized state, as demonstrated by increased TNFα secretion and decreased levels of DC-SIGN and M2-associated IL-1017. Co-culturing naïve primary macrophages with MSCs stimulated CD86 expression, attributing a shift towards M1-like macrophages17. Moreover, the study also showed that the co-culture of MSCs with M1-like macrophages led to a reduction in inflammatory M1-like macrophages, accompanied by a shift towards M2-like macrophages17. Conversely, the co-culture of MSCs with M2-like macrophages further enhanced the M2 polarization14. Vasandan et al. concluded that MSC-secreted factors at the MSC-macrophage interface repolarize macrophages by modifying metabolic patterns in variably polarised macrophages17. To date, there has been a strong focus on identifying TAMs in the 'either/or' M1 or M2 state, sometimes disregarding other theories10. A new study found the expression of both M1 and M2 genes in identical cells18. Interestingly, Rabani et al. discovered that when MSCs were exposed to M0 macrophages, they produced both M1-like and M2-like macrophage phenotypes simultaneously18. From the study, M2-like macrophages showed elongated morphology, were CD163+, had acute phagosomal acidification, low NOX2 expression, and limited phagosomal superoxide production while the M1-like macrophages produced high levels of phagosomal superoxide but with low or undetectable levels of CD4018. Prostaglandin E2 and phosphatidylinositol 3-kinase were essential for promoting the M1-like macrophages18. Because TAMs are highly abundant in tumors, more research is necessary to understand how MSC and macrophages interact to regulate the activation state of macrophages, which will aid in the development of an effective therapeutic strategy for breast cancer.
The present study aims to repolarize M1-like macrophages within the BC microenvironment by co-culturing UC-MSCs with M0 macrophages. Subsequently, the influence of the M0 macrophages co-cultured with UC-MSCs was explored by treating MDA-MB-231 cells with conditioned medium from the co-culture of UC-MSCs with M0 macrophages. In parallel, M0 macrophages treated with MSCs were also cultured directly with MDA-MB-231 to evaluate their effect on TNBC cell growth and their mechanism of action. Repolarizing M1-like macrophages with MSCs may enhance anti-tumor responses and inhibit TAMs, which exhibit pro-tumor responses in the BC microenvironment.
Methods
Materials
Human umbilical cord mesenchymal stem cells (UC-MSCs), human THP-1 cells, and the MDA-MB-231 cell line were procured from the American Type Culture Collection (ATCC, USA). Roswell Park Memorial Institute (RPMI)-1640 medium, Dulbecco's Modified Eagle Medium/F12 (DMEM/F12), fetal bovine serum (FBS), penicillin and streptomycin (Pen-Strep), phorbol 12-myristate 13-acetate (PMA), lipopolysaccharide (LPS), and phosphate-buffered saline (PBS) were secured from Thermo Fisher Scientific (USA).
Culturing UC-MSCs, THP-1 cells, and MDA-MB-231 cells
UC-MSCs were cultured in DMEM/F12 media containing L-glutamine and sodium bicarbonate, 10% FBS, and 1% Pen-Strep in a 37°C incubator with 5% CO2. UC-MSCs between passages 7–10 were used for all the experiments. THP-1 cells were grown in RPMI1640 media containing L-glutamine and supplemented with 10% FBS and 1% Pen-Strep. THP-1 cells were incubated with 20 nM PMA for 48 hours to differentiate them into naïve macrophages (M0). The influence of UC-MSCs on M0 macrophage differentiation into M1 and M2 macrophages was assessed by culturing both cell types together in 6-well culture plates (CORNING, USA), physically separated by cell-culture inserts (0.4 microns, CORNING, USA) for 30 hours. 3.5 × 105 THP-1 cells were used for differentiation experiments, and 3.5 × 104 MSCs were seeded on culture inserts in co-culture conditions17. The ratio was established by considering the proportions of MSCs inducing immune suppression in MLRs, as previously reported by Vasandan et al.17. The respective conditions for co-culture of UC-MSCs with M0 macrophages were untreated M0 macrophages, LPS-treated M0 macrophages, UC-MSCs co-cultured with M0 macrophages, and UC-MSCs co-cultured with LPS-treated M0 macrophages for 30 hours. The conditioned medium (CM) from respective treatments was used to treat MDA-MB-231 cells for 72 hours. Meanwhile, the cells from respective treatments were directly co-cultured with MDA-MB-231 cells for 72 hours.
UC-MSCs Immunophenotyping
UC-MSCs and UC-MSCs co-cultured with M0 macrophages for 30 hours with respective treatments were evaluated for their surface markers using the BD Stemflow™ Human MSC Analysis Kit (BD Bioscience, USA) based on the manufacturer's instructions. Briefly, both types of UC-MSCs were suspended in a tube containing at least 1 × 105 cells each in 100 µl of BD Pharmingen™ stain buffer. The cells were mixed with the appropriate monoclonal antibodies for anti-human CD105 (conjugated with PerCP-Cy™5.5), CD73 (conjugated with APC), CD90 (conjugated with FITC), CD19, CD11beta, CD45, CD34, and HLA-DR (all negative markers conjugated with PE) or the respective isotype control. Cells stained with isotype control from the kit and unstained cell suspension were used as controls. The antibodies were included at concentrations recommended by the manufacturers, and the samples were left to incubate away from light for 30 minutes at 2–8 ℃. The samples were then washed twice with PBS and analyzed on a NovoCyte Advanteon Flow Cytometer (Agilent, USA), recording at least 10,000 events per sample. The obtained data were further analyzed using NovoExpress software.
Enzyme-Linked Immunosorbent Assay (ELISA)
TNF-α and IL-10 were quantified in the supernatants of untreated and respective treated M0 macrophages at each treatment time using the TNF-α and IL-10 DuoSet ELISA kit (R&D Systems, USA) according to the guidelines stated by the manufacturer. Firstly, a 96-well ELISA plate was coated with either TNF-α or IL-10 capture antibodies, respectively, and incubated overnight at 4°C. The plate was washed with wash buffer (400 µl) three times. Subsequently, diluent buffer was added to block the plate and incubated at room temperature for one hour. The blocking buffer was pipetted out, and the washing step was repeated as previously. Standards were prepared by diluting them in reagent diluent for a total of seven points, and both samples and diluted standards (100 µl) were added into respective wells of the plate. The plate was sealed properly and incubated overnight at 4°C. The plate was washed with wash buffer three times post-incubation. Next, TNF-α or IL-10 detection antibodies (100 µl) were added and incubated for 2 hours at room temperature. The detection antibodies were then aspirated, and the washing step was repeated. Subsequently, Streptavidin-HRP (100 µl) was added to each well, and the plate was left at room temperature for a 20-minute incubation period. The aspiration and washing steps were repeated. Finally, substrate solution (100 µl) was added to each well and then incubated for 20 minutes at room temperature. The reaction was terminated by adding stop solution (50 µl) and the optical density of each well was evaluated immediately using a FLUOstar Omega microplate reader (BMG Labtech, Germany) set to 450 nm and 570 nm for wavelength correction.
MTT Assay
The anti-cancer activity of both direct cell-cell interaction and CM of M0 macrophages co-cultured with UC-MSCs (M0/MSCs) on MDA-MB-231 cells was determined by the MTT (3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide) assay. For the effect of CM, MDA-MB-231 cells (5 × 103/well) were plated in 96-well plates 24 hours before treatment. During treatment, the old medium from the wells was removed and replaced with CM from M0/MSCs (100 µl). For direct cell-cell effect, MDA-MB-231 cells (5 × 103/well) and M0/MSCs cells (5 × 103/well) were seeded together in the well. Both MDA-MB-231 cells for either CM or direct cell-cell treatment were incubated at 24, 48, and 72 hours. Post-treatment, the old media was removed and new media (90 µl) and MTT (10 µl) were added into each well. The MTT assay (Invitrogen, USA) was initially prepared by dissolving in PBS at 5 mg/ml. The plates were incubated at 37°C with 5% CO2 for 3 hours and the medium was then replaced with 100 μL DMSO. The plates were then incubated for 10 minutes at 37°C with 5% CO2 and the absorbance for each well was measured at 570 nm using an ELISA microplate reader.
Total RNA Extraction and cDNA synthesis
Total RNA was extracted from MDA-MB-231 cells cultured with CM of M0/MSCs (in a ratio of 1:1 with fresh culture media) and MDA-MB-231 cells that directly co-cultured with M0/MSCs for 72 hours using TRIsure™ (Bioline, UK) following the instructions stated by the manufacturer. NanoDrop (Thermo Fisher Scientific, USA) was used to evaluate the concentration and purity of RNA with an A260:A280 ratio of 1.7–1.9 obtained, demonstrating isolated samples were high-quality RNA with low levels of protein contamination. RNA integrity was assessed by conducting agarose gel electrophoresis with the presence of 28S and 18S rRNA bands. Then, RNA (2 μg) was reverse transcribed into cDNA using the Tetro cDNA Synthesis kit (Bioline, UK) based on instructions stated by the manufacturer. Briefly, the reaction conditions were as follows: 2 μg of RNA, 4 μL of 5x RT buffer, 1 μL of Oligo (dT)18 primer mix, 1 μL of dNTP mix 10 nM, 1 μL of RNase inhibitor, 1 μL of (200 U/μL) reverse transcriptase, and DEPC water to a final volume of 20 μL. Samples were mixed and placed at 45°C for 30 minutes incubation, followed by incubating at 85°C for 5 minutes to terminate the reaction and chilled on ice.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Relative mRNA expression levels were detected via qPCR assays on a StepOnePlus Real-Time PCR System (Applied Biosystems, USA). All PCR reactions were carried out in triplicates using a SensiFAST SYBR Hi-Rox Kit (Bioline, UK). Reactions were set up in a final volume of 20 μL containing: 10 μL 2x SensiFAST SYBR® Hi-ROX Mix, 0.8 μL of each primer pair mixture [10 μM of each primer], 2 μL of cDNA and 6.4 μL of nuclease-free water to make up the total volume. All primer sequences are displayed in Table 1. The three-step cycling conditions included an initial polymerase activation at 95°C for 2 minutes, followed by 40 cycles of denaturation at 95°C for 5 seconds, annealing, and extension at 60°C for 10 seconds and 75°C for 5 seconds, respectively. The assessment of mRNA expression levels relative to GAPDH, used as the reference gene, was executed through the ΔΔCt method. To achieve relative quantification, we employed the 2−ΔΔCt method.
| Gene | Forward (5’-3’) | Reverse (5’-3’) |
| GAPDH | AATCCCATCACCATCTTCCA | TGGACTCCACGACGTACTCA |
| CD80 | CACTTCTGTTCAGGTGTTATCC | GGTGTAGGGAAGTCAGCTTTG |
| CD163 | ACATAGATCATGCATCTGTCATTTG | CATTCTCCTTGGAATCTCACTTCTA |
| IRF-5 | TTCTCTCCTGGGCTGTCTCTG | CTATACAGCTAGGCCCCAGGG |
| IRF-4 | GCTGATCGACCAGATCGACAG | CGGTTGTAGTCCTGCTTGC |
| TNF-α | CCTCTCTCTAATCAGCCCTCTG | GAGGACCTGGGAGTAGATGAG |
| IL-10 | TACGGCGCTCGTCATCGATTT | TAGAGTCGCCACCCTGATGT |
| Akt1 | GAAGGACGGGAGCAGGCGGC | CCTCCTCCAGGCAGCCCT |
| mTOR | AGTGGACCAGTGGAAACAGG | TTCAGCGATGTCTTGTGAGG |
| YKL-39 | AAGATGACCTTGCTGCCT | TGATCTAAGAGGAAGTCAGG |
| P53 | CCCCTCCATCCTTTCTTCTC | ATGAGCCAGATCAGGGACTG |
Statistical Analysis
For all conducted experiments, the reported values are the means derived from a minimum of three distinct trials, along with their corresponding standard deviations (SD), unless specified differently. Every experiment was replicated in triplicate. To compare data among different treatment conditions and test samples, we employed a Student's independent t-test. In each experiment, pairwise comparisons were conducted between two groups at a time. Statistical significance was determined at *P < 0.05, **P < 0.01, and ***P < 0.001.






Results
UC-MSCs co-cultured with M0 macrophages and LPS-treated macrophages displayed a low percentage of CD105 expression
UC-MSCs showed strong expression of CD90 (> 98%) and CD73 (> 98%) but low expression of CD105 and no expression of any negative MSC markers when co-cultured with M0 macrophages, LPS-treated M0 macrophages, and UC-MSCs alone (Figure 1A-C).
UC-MSCs and LPS transform M0 macrophages into an elongated shape
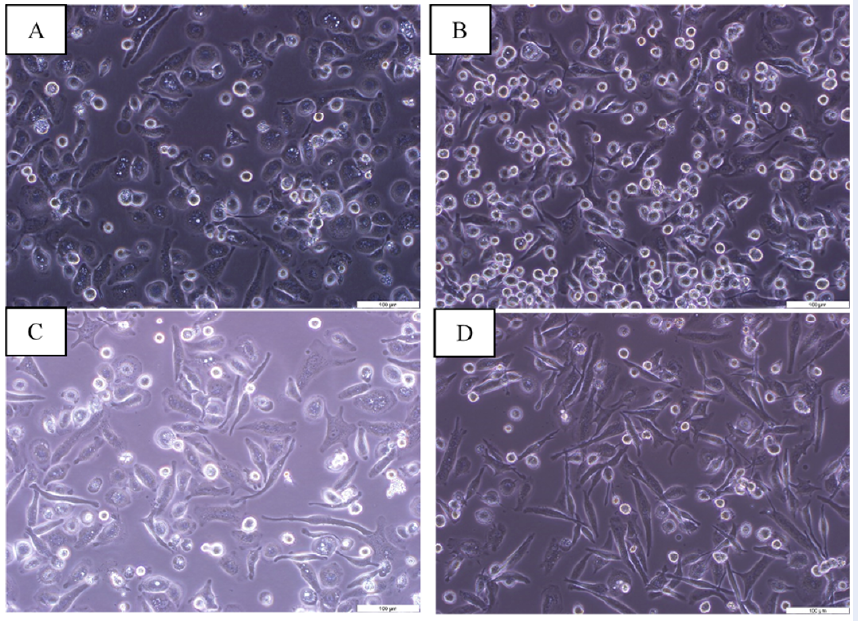
M0 macrophages adhered to the cell culture plate and had outstretched parapodia (Figure 2A), while LPS-treated M0 macrophages acquired a rounded and flat shape phenotype (Figure 2B). M0 macrophages and LPS-treated M0 macrophages transformed their round shape to an elongated shape when co-cultured with UC-MSCs, respectively (Figure 2C-D).
M0 macrophages co-cultured with UC-MSCs exhibited lower expression of CD163, TNF-α, and IL-10 genes but induced IRF-5 gene expression
LPS-treated M0 macrophages co-cultured with UC-MSCs significantly increased CD80 gene expression compared to untreated M0 macrophages (P < 0.05) (Figure 3A). M0 macrophages co-cultured with UC-MSCs significantly inhibited CD163 gene expression compared to untreated M0 macrophages (P < 0.001) (Figure 3B). Similarly, M0 macrophages and LPS-treated M0 macrophages co-cultured with UC-MSCs inhibited TNF-α gene expression compared to untreated M0 macrophages (P < 0.01 and P < 0.001, respectively). In addition, LPS-treated M0 macrophages co-cultured with UC-MSCs also suppressed TNF-α gene expression compared to LPS-treated M0 macrophages (P < 0.01) (Figure 3C). Furthermore, M0 macrophages co-cultured with UC-MSCs reduced IL-10 gene expression compared to untreated M0 macrophages (P < 0.001) (Figure 3D). On the other hand, LPS-treated M0 macrophages co-cultured with UC-MSCs significantly increased IRF-4 gene expression when compared with untreated M0 macrophages and LPS-treated M0 macrophages (P < 0.05) (Figure 3E). However, only M0 macrophages co-cultured with UC-MSCs significantly induced IRF-5 gene expression compared to untreated M0 macrophages (P < 0.05) (Figure 3F).
CM from UC-MSCs co-cultured with M0 macrophages and LPS-treated macrophages did not contain TNF-α
CM from UC-MSCs co-cultured with M0 macrophages and LPS-treated macrophages did not contain TNF-α (Figure 4A). However, CM from LPS-treated M0 macrophages significantly highly secreted TNF-α compared to CM from M0 macrophages (P < 0.001) (Figure 4A). Meanwhile, CM from LPS-treated M0 macrophages and CM from UC-MSCs co-cultured with M0 macrophages and LPS-treated macrophages highly secreted IL-10 compared to CM from M0 macrophages, but not statistically significantly (Figure 4B).
The proliferation of MDA-MB-231 cells was suppressed when cultured with CM from M0 macrophages co-cultured with UC-MSCs through the inhibition of AKT1 and YKL-39 genes
CM from M0 macrophages, CM from M0 macrophages co-cultured with UC-MSCs, and CM from LPS-treated M0 macrophages co-cultured with UC-MSCs significantly inhibited the proliferation of MDA-MB-231 cells at 72 hours compared to 24 hours (P < 0.05) (Figure 5A). The expression of the AKT1 gene was reduced in MDA-MB-231 cultured with CM from M0 macrophages and CM from M0 macrophages co-cultured with UC-MSCs compared to untreated MDA-MB-231 (control) (P < 0.001 and P < 0.05 respectively) (Figure 5B). However, the expression level of AKT1 was increased in MDA-MB-231 cultured with CM from M0 macrophages co-cultured with UC-MSCs, CM from LPS-treated M0 macrophages, and CM from LPS-treated M0 macrophages co-cultured with UC-MSCs compared to MDA-MB-231 cultured with CM from M0 macrophages (P < 0.01 and P < 0.05 respectively) (Figure 5B). Furthermore, the p53 gene expression level was significantly decreased in MDA-MB-231 cultured with CM from M0 macrophages (P < 0.001) (Figure 5C). Meanwhile, MDA-MB-231 cultured with CM from M0 macrophages co-cultured with UC-MSCs and CM from LPS-treated M0 macrophages displayed a significantly higher level of p53 gene expression compared to MDA-MB-231 cultured with CM from M0 macrophages (P < 0.01) (Figure 5C). On the other hand, there are no significant differences in mTOR gene expression in MDA-MB-231 cultured with CM from all respective treatments (Figure 5D). In addition, the expression level of the YKL-39 gene was reduced in MDA-MB-231 cultured with CM from M0 macrophages and CM from M0 macrophages co-cultured with UC-MSCs compared to untreated MDA-MB-231 (control) (P < 0.001 and P < 0.01) (Figure 5E).
MDA-MB-231 cells directly co-cultured with M0 macrophages co-cultured with UC-MSCs displayed a consistent increase in cell proliferation
The results of the cell viability study indicated that MDA-MB-231 co-cultured with M0 macrophages, M0 macrophages co-cultured with UC-MSCs, LPS-treated M0 macrophages, and LPS-treated M0 macrophages co-cultured with UC-MSCs consistently increase in cell proliferation compared to the control group at both 24 and 48 hours (Figure 6A). However, as the study progresses to the 72-hour mark, a slight deviation from this trend is observed when MDA-MB-231 is co-cultured with LPS-treated M0 macrophages and LPS-treated M0 macrophages co-cultured with UC-MSCs (P < 0.05) (Figure 6A). However, the treatments still maintain a relatively high level of cell viability, with only a minor decrease of approximately 5% below the baseline 100% viability observed when MDA-MB-231 is co-cultured with LPS-treated M0 macrophages co-cultured with UC-MSCs (Figure 6A). At 72 hours, the expression level of the AKT1 gene was significantly reduced in MDA-MB-231 co-cultured with LPS-treated M0 macrophages co-cultured with UC-MSCs compared to untreated MDA-MB-231 (control) (P < 0.01) (Figure 6B). There are no significant differences in p53 gene expression in all MDA-MB-231 treated groups (Figure 6C). Meanwhile, MDA-MB-231 co-cultured with M0 macrophages, M0 macrophages co-cultured with UC-MSCs, LPS-treated M0 macrophages, and LPS-treated M0 macrophages co-cultured with UC-MSCs had a significantly lower expression of the mTOR gene compared to untreated MDA-MB-231 (control) (P < 0.05, P < 0.001, P < 0.01) (Figure 6D). In addition, the expression level of the YKL-39 gene was significantly reduced in MDA-MB-231 co-cultured with M0 macrophages and M0 macrophages co-cultured with UC-MSCs compared to untreated MDA-MB-231 (control) (P < 0.05 and P < 0.01) (Figure 6E). In contrast, MDA-MB-231 co-cultured with LPS-treated M0 macrophages co-cultured with UC-MSCs had a significantly higher expression of the YKL-39 gene compared to MDA-MB-231 co-cultured with M0 macrophages (P < 0.05) (Figure 6E).
Discussion
Mesenchymal stem cells, also known as multipotent stromal cells (MSCs), represent a subset of non-hematopoietic adult stem cells that can be found in a wide range of tissues throughout the body19. They play a crucial role as resident tissue reservoirs for precursor cells, facilitating tissue replacement and repair through their unique capacity for differentiation and their ability to influence the adjacent surroundings by secreting trophic factors20. The phenotype of the UC-MSCs employed in our current study was characterized by positive expression of CD73, CD90, and CD105, while being negative for CD34, CD45, CD11b, CD14, CD19, CD79α, and HLA-DR. These findings are consistent with previous observations, even when these cells were co-cultured with M0 macrophages and LPS-treated M0 macrophages for a duration of 30 hours. This characterization aligns with the guidelines established by the International Society for Cellular Therapy (ISCT)21. However, it is worth noting that the percentage of CD105-positive cells ranged from 26% to 35%. While CD105 is commonly considered a signature MSC marker, research addressing its applicability is not entirely consistent. For instance, Cleary et al. demonstrated that the chondrogenic differentiation potential of bone marrow-derived MSCs was not dependent on their CD105 surface expression22. Dizaji et al. reported that 76% of amniotic membrane-derived MSCs were CD105-positive, while 92% of adipose tissue-derived MSCs exhibited this marker23. Lee et al. demonstrated that the culture conditions may control the expression of CD105 in MSCs generated from bone marrow, with alginate and transforming growth factor beta-3 significantly lowering its expression24. As a result, the origin of the UC-MSCs, the number of passages, and the particular culture conditions used could all have an impact on the percentage of CD105 surface expression (< 90%) for our study.
The TME in breast cancer consists of a population of tumor cells that interact with a variety of immune cells and MSCs, creating a complex network driving tumor development25, 26. Apart from their direct interaction with tumor cells, MSCs regulate different types of immune cells in the TME, such as neutrophils, macrophages, and natural killer cells, which eventually affects the growth of the tumor. Macrophages are heterogeneous cells that can be classified as either M1-like or M2-like macrophages based on the particular stimuli present at the time of activation. Notably, TAMs isolated from metastatic tumors often display a suppressive M2-like phenotype. To close the existing gaps in our understanding of the mechanisms at the MSC-macrophage interface in breast cancer, this current work aims to analyze the interactions between MSCs and macrophages at different activation states. THP-1 monocytes, once differentiated, are often used as an in vitro model for human macrophages. From these THP-1 cells, M1-like and M2-like macrophage populations were generated through co-culturing with UC-MSCs. Microscopic examination revealed that PMA-treated THP-1 cells (M0 macrophages), LPS-treated M0 macrophages, M0 macrophages co-cultured with UC-MSCs, and LPS-treated M0 macrophages co-cultured with UC-MSCs all exhibited adhesion characteristics, displaying the typical flattened, amoeboid-like, elongated, and branching macrophage morphology. The assessment of M0 macrophages co-cultured with M0 macrophages were treated with UC-MSCs in the absence of M1 or M2 stimuli (IFN-γ and IL-4, respectively) to observe if they could shift towards an M1 or M2-like phenotype. LPS without IFN-γ has been found to bind Toll-like receptor 4 on the surface of M0 macrophages, facilitating their transition to an M1-like phenotype27. Peshkova et al. previously selected UC-MSCs for their study due to high levels of cytokines identified in their conditioned medium compared to MSCs from adipose tissue, bone marrow, gingival tissue, and placenta28. Pro-inflammatory cytokines such as IFNγ, TNF-α, IL-1β, and IL-6 made up the majority of the cytokines detected in the UC-MSCs conditioned media from the study, whereas anti-inflammatory cytokines such as IL-1RA, IL-10, and IL-13 were found at lower quantities28. It is well established that MSCs have an immunomodulatory influence on macrophage polarization away from traditional pro-inflammatory M1-like macrophages and toward alternatively activated anti-inflammatory M2-like macrophages. However, the mechanism of MSC-mediated macrophage phenotypic regulation is currently still being investigated28. Our work indicated a substantial increase in IRF-4 expression in LPS-treated macrophages co-cultured with UC-MSCs compared to those without UC-MSCs. This is consistent with earlier studies because IRF-4 is a transcription factor that drives M2 activation29. In naive or M0 macrophages, UC-MSCs skewed the macrophages toward an M1-like phenotype by enhancing the expression of the IRF-5 gene while suppressing the CD163 and IL-10 genes, both of which are associated with M2-like macrophages. UC-MSCs also inhibited the expression of the TNF-α gene, a marker of M1-like macrophages, in both M0 macrophages and LPS-treated M0 macrophages. This finding aligns with previous research demonstrating the suppression of TNF-α secretion by MSC-conditioned media in M1-like macrophages30, 31. As a result, our findings demonstrate that UC-MSCs can exert immunomodulatory effects via soluble factors, effectively converting M0 macrophages to an M1-like phenotype.
MSCs produced from umbilical cord/cord blood (UC-MSCs) have been shown in studies to have better anti-cancer capability, inhibiting the growth of solid tumor cell lines such as breast, liver, prostate, and bladder cancer. BM-MSCs mostly promote tumor growth, while some studies have found anti-proliferative effects32. We then investigated the interaction between M0 macrophages co-cultured with UC-MSCs and MDA-MB-231 cells, a TNBC cell line. To investigate this interaction, we treated MDA-MB-231 cells both directly via cell-to-cell contact and indirectly utilizing conditioned media (CM) from M0 macrophages co-cultured with UC-MSCs (M0/MSCs). A study investigated the effects of CM from THP-1 macrophages differentiated with PMA and human M1 macrophages on the colon cancer cell lines HT29 and CACO-233. The study found G0/G1 and G2/M cell cycle arrest, but the influence of TNF-α and CXCL9 generated by M1-like macrophages on HT-29 cell proliferation appeared insignificant. This suggests the participation of other macrophage-released mediators in lowering cancer cell proliferation33. Another study by Song et al. reported that 25% CM from naive macrophages could restrain the proliferation of both MCF-7 and MDA-MB-231 breast cancer cell lines. Inflammatory cytokines, particularly IL-6 and IL-8, were implicated in limiting breast cancer cell growth rather than promoting it34. In our investigation, CM from M0 macrophages, along with CM from M0 macrophages co-cultured with UC-MSCs and CM from LPS-treated M0 macrophages co-cultured with UC-MSCs, significantly inhibited MDA-MB-231 cell proliferation at 72 hours compared to their respective treatments at 24 hours. Similar inhibitory patterns were observed in MDA-MB-231 cell proliferation when co-cultured with LPS-treated macrophages, although statistical significance was not achieved. Additionally, CM from human bone marrow MSCs (BM-MSCs) previously inhibited the proliferation of CAL-27 and FaDu squamous cell carcinoma cell lines by lowering the expression of PCNA (a proliferation marker) and BCL-2 (an anti-apoptotic protein) in both cell lines35. CM from Wharton's jelly-derived MSCs exhibited potent inhibition of proliferation in T47D and MCF-7 breast cancer cell lines36. Khalil et al. also demonstrated the anti-proliferative and anti-apoptotic effects of MSCs from adipose tissue, bone marrow, and umbilical cord on ovarian cancer cell lines, leading to a significant reduction in cancer markers and cell proliferation37. CM from M0/UC-MSCs with or without LPS may contain multiple secretomes, which can either positively or negatively affect cell behavior38. Interferon-β (IFN-β) secretion by UC-MSCs has been demonstrated to effectively impede MDA-MB-231 cell proliferation by triggering apoptosis39. Furthermore, it is possible that UC-MSCs may secrete exosomes within the CM of M0/UC-MSCs. Exosomes are nanosized membrane vesicles ranging from 40 to 160 nm in diameter, which can be released by various cell types40. These exosomes contain a complex cargo, including nucleic acids such as DNA, mRNAs, and noncoding RNAs, lipids, and a variety of proteins41. Remarkably, multiple studies have highlighted the central role of MSC-derived exosomes in modulating TME and influencing the development of various human cancers42. Moreover, MSC-derived exosomes have demonstrated anti-angiogenic properties. In a breast cancer model, Lee et al. demonstrated that exosomes derived from MSCs could suppress angiogenesis by downregulating the expression of VEGF43. Zhou et al. have provided evidence that exosomes secreted from bone marrow-derived MSCs can accelerate anti-tumoral macrophage polarization and facilitate the engagement of cytotoxic lymphocytes, thereby improving immune-based treatment for pancreatic cancer within a living organism44. As a result, it is possible that unidentified pro-inflammatory substances, such as cytokines or exosomes from M0 macrophages, may become more abundant after co-culture with UC-MSCs, leading to their anti-cancer action.
Regarding direct interactions between M0 macrophages and MDA-MB-231 cells, noteworthy inhibitory effects were primarily observed in LPS-treated macrophages, both with and without co-culture alongside UC-MSCs, at the 72-hour time point in comparison to their 48-hour counterparts. However, it's important to highlight that the percentages of cell viability for both cell types when co-cultured directly with MDA-MB-231 cells mostly remained higher compared to the viability of the control group. In the control group, MDA-MB-231 cells were cultured alone without any treatment for 24 hours, 48 hours, and 72 hours, respectively. This suggests that direct cell-to-cell contact may potentially influence macrophages to support the TNBC cell line growth rather than inhibiting it, which contrasts with the outcomes observed when using CM. Interestingly, a previous study demonstrated that direct mixed co-cultures of M1 and M2 macrophages could enhance the proliferation of T98G cells, a human glioblastoma cell line, with M2 macrophages promoting significantly higher proliferation rates45. Direct physical contact between macrophages and tumor cells also provided protection to the tumor cells against apoptosis induced by chemotherapy drugs, whereas interactions between these cells without direct contact did not yield the same effect46. Furthermore, this direct co-culture could result in greater STAT3 activation in tumor cells when compared to indirect co-culture when physically separated using a transwell co-culture system47. An earlier study indicated that the activation of STAT3 in ovarian and kidney cancer cells significantly increased when these cells were directly co-cultured with macrophages48, 49. Macrophages may have a supporting role in promoting cancer cell proliferation and survival in patients with malignant tumors, as STAT3 activation is intimately associated with these two outcomes50. Macrophages and cancer cells' direct interaction promote the growth of the cancer cells, possibly regulated by STAT3.
The following intriguing question concerned the genes involved in regulating the reduction in MDA-MB-231 cell proliferation when co-cultured with CM generated from M0/MSCs. Notable effects were only observed on the expression of AKT1 and YKL-39 genes in MDA-MB-231 cells when treated with CM from M0 macrophages co-cultured with UC-MSCs, as compared to the control group. The role of AKT1 genes in breast cancer involves promoting cell survival and proliferation through its involvement in the PI3K-AKT-mTOR signaling pathway51. In a study by Choi et al. the anti-cancer effects of compound K (CK) were examined in SKBR3 and MDA-MB-231 cells, revealing that CK hinders cancer cell proliferation and induces apoptosis by targeting AKT151. Additionally, AKT activation has been reported to trigger resistance to tamoxifen in breast cancer cells52, 53. Arranz and colleagues have highlighted the significant involvement of AKT isoforms in macrophage polarization in which the absence of the AKT1 isoform promotes the M1-like phenotype, whereas the loss of the AKT2 isoform leads to the development of the M2-like phenotype54. This observation suggests that the CM from M0/MSCs may contain a high level of M1-like macrophage-associated secretomes, which are responsible for suppressing AKT1 gene expression. On the other hand, YKL-39, a closely related homolog of YKL-40, was initially identified as a highly secreted protein in primary cultures of human articular chondrocytes55. Initially, YKL-39 was proposed as a biomarker for chondrocyte activation and the progression of osteoarthritis in humans56. The only reported study investigating the link between YKL-39 and cancer was conducted by Kavsan et al., who revealed heightened CHI3L2 gene expression in glioblastoma57. Subsequently, Liu et al. conducted a study that revealed YKL-39 expression in TAMs but not in human breast cancer cells58. Therefore, the suppression of MDA-MB-231 cell growth following culture with CM from M0/MSCs may result in a reduced TAMs population, leading to the inhibition of the YKL-39 gene.
Our current work will undoubtedly benefit from a number of improvements that could improve understanding of the MSC-macrophage-breast cancer cell interaction and assist to design more effective therapeutic options targeting the TME. Our study employed two-dimensional (2D) cell culture which is the most common research model utilized since the early 1900s. Future study can take advantage of the 3D cell culture technology, which has recently gained popularity for various applications, particularly in cancer research, stem cell research, pharmaceutical development, and disease investigations59. When it comes to simulating the biological behavior of tumor cells, particularly the mechanisms that lead to therapeutic escape and drug resistance, the 2D culture technique is appearing less appropriate than the 3D cell culture approach60. However, although these 3D models are promising, their application still presents several obstacles. The selection of the most appropriate 3D model to the question posed is also based on the analytical processes that will be done60. With 3D culture, there is still the need to break up spheroids into a single-cell suspension prior to subsequent assay analysis, as well as a limitation due to the automation of liquid handling when using viscous liquids such as collagen- and Matrigel-based hydrogels59. Since our in vitro study exposed macrophages to MSCs for 30 hours, which may not precisely reflect interactions in vivo, more analysis is required to understand the timing of the effect18. Moreover, MSCs from other sources, such as bone marrow or adipose tissue, may exhibit different properties. Additionally, MSCs can vary between donors, potentially influencing their interactions with tumor cells and their immunomodulatory effects. The current study employs CM to investigate indirect cell interactions but does not fully characterize the composition of these media. Identifying and quantifying the various factors present, such as cytokines, chemokines, growth factors, and exosomes, is crucial for elucidating the mechanisms driving the observed effects. Furthermore, the current research was based on analyses at specific time points, which might not capture the full dynamics of cell interactions and gene expression changes. More frequent sampling could provide insight into transient or early signaling events. The reliance on gene expression data alone to infer functional outcomes might overlook important aspects of cellular responses. Study on protein expression levels and conducting functional assays such as migration and invasion assays for cancer cells or phagocytosis assays for macrophage would offer a more holistic view of cellular activities. Lastly, while the study identifies gene expression changes linked to MSC-macrophage interactions and their impact on breast cancer cells, it lacks a thorough investigation of the underlying signaling pathways and molecular mechanisms. A deeper exploration into these aspects in future study could enhance our understanding of the MSC-macrophage-breast cancer cell interactions and contribute to the development of more effective TME-targeted therapies.
Conclusions
Our research reveals that UC-MSCs are capable of inducing an M1-like phenotype in M0 macrophages. The CM from these M0 macrophages, which have been influenced by MSCs, may contain anti-tumor factors derived from both M1 macrophages and MSCs. These factors have the potential to inhibit the growth of a TNBC cell line by targeting key molecules such as AKT1 and YKL-39. This study highlights the need for further investigation into the interactions between MSCs, macrophages, and BC cells. Such research could lead to the development of more effective therapeutic strategies aimed at targeting the TME to enhance TNBC treatment outcomes.
Abbreviations
AKT1 - A serine/threonine-specific protein kinase, BC - Breast Cancer, BCL-2 - B-cell lymphoma 2, BM-MSCs - Bone Marrow Mesenchymal Stem Cells, CD - Cluster of Differentiation, CM - Conditioned Medium, CTLs - Cytotoxic T Lymphocytes, CXCL9 - Chemokine (C-X-C motif) ligand 9, DEPC - Diethylpyrocarbonate, DMEM - Dulbecco's Modified Eagle Medium, ELISA - Enzyme-Linked Immunosorbent Assay, FBS - Fetal Bovine Serum, HLA-DR - Human Leukocyte Antigen - DR isotype, IFNγ - Interferon gamma, IL - Interleukin, ISCT - International Society for Cellular Therapy, IRF - Interferon Regulatory Factor, LPS - Lipopolysaccharide, M0 - Naïve macrophages, M1 - Classically activated macrophages, M2 - Alternatively activated macrophages, MDA-MB-231 - A type of human breast cancer cell line, MSC - Mesenchymal Stem Cells, MTT - 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyl tetrazolium bromide, mTOR - Mammalian Target of Rapamycin, NOX2 - NADPH oxidase 2, PBS - Phosphate-Buffered Saline, PCNA - Proliferating Cell Nuclear Antigen, PGE2 - Prostaglandin E2, PMA - Phorbol 12-myristate 13-acetate, qRT-PCR - Quantitative Real-Time Polymerase Chain Reaction, RPMI - Roswell Park Memorial Institute Medium, SD - Standard Deviation, STAT3 - Signal Transducer and Activator of Transcription 3, TAM - Tumor-Associated Macrophages, TME - Tumor Microenvironment, TNBC - Triple-Negative Breast Cancer, TNF-α - Tumor Necrosis Factor-alpha, UC-MSCs - Umbilical Cord Mesenchymal Stem Cells, VEGF - Vascular Endothelial Growth Factor, YKL-39 - A gene/protein sometimes associated with inflammation
Acknowledgments
None.
Author’s contributions
RM, MAY, BHY, MYA, MSR designed the research study. NRJ performed experimental works. RM and NRJ wrote the manuscript. RM is the principal investigator for Research University Individual and Special Research grant that funded this research. All author read and approved the final version of manuscript for submission.
Funding
Research University Individual grant, Universiti Sains Malaysia (1001/CIPPT/8012328) and Special Research University grant by Universiti Sains Malaysia for BCTRP (1001/CIPPT/8070033) that fund this research.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Arnold
M.,
Morgan
E.,
Rumgay
H.,
Mafra
A.,
Singh
D.,
Laversanne
M.,
Current and future burden of breast cancer: global statistics for 2020 and 2040. The Breast.
2022;
66
:
15-23
.
View Article Google Scholar -
Arneth
B.,
Tumor microenvironment. Medicina (Kaunas, Lithuania).
2019;
56
(1)
:
15
.
View Article Google Scholar -
Belli
C.,
Trapani
D.,
Viale
G.,
D'Amico
P.,
Duso
B.A.,
Della Vigna
P.,
Targeting the microenvironment in solid tumors. Cancer Treatment Reviews.
2018;
65
:
22-32
.
View Article Google Scholar -
Qiu
S.Q.,
Waaijer
S.J.,
Zwager
M.C.,
de Vries
E.G.,
van der Vegt
B.,
Schröder
C.P.,
Tumor-associated macrophages in breast cancer: innocent bystander or important player?. Cancer Treatment Reviews.
2018;
70
:
178-89
.
View Article Google Scholar -
Gao
Y.,
Bado
I.,
Wang
H.,
Zhang
W.,
Rosen
J.M.,
Zhang
X.H.,
Metastasis organotropism: redefining the congenial soil. Developmental Cell.
2019;
49
(3)
:
375-91
.
View Article Google Scholar -
DeNardo
D.G.,
Ruffell
B.,
Macrophages as regulators of tumour immunity and immunotherapy. Nature Reviews. Immunology.
2019;
19
(6)
:
369-82
.
View Article Google Scholar -
Hourani
T.,
Holden
J.A.,
Li
W.,
Lenzo
J.C.,
Hadjigol
S.,
O'Brien-Simpson
N.M.,
Tumor associated macrophages: Origin, recruitment, phenotypic diversity, and targeting. Frontiers in Oncology.
2021;
11
:
788365
.
View Article Google Scholar -
Lu
J.,
Ma
L.,
The role of tumor-associated macrophages in the development, metastasis and treatment of breast cancer. Pathology-Research and Practice.
2020;
216
(9)
:
153085
.
View Article Google Scholar -
Chen
Y.,
Song
Y.,
Du
W.,
Gong
L.,
Chang
H.,
Zou
Z.,
Tumor-associated macrophages: an accomplice in solid tumor progression. Journal of Biomedical Science.
2019;
26
(1)
:
78
.
View Article Google Scholar -
Kowal
J.,
Kornete
M.,
Joyce
J.A.,
Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy.
2019;
11
(8)
:
677-89
.
View Article Google Scholar -
Hmadcha
A.,
Martin-Montalvo
A.,
Gauthier
B.R.,
Soria
B.,
Capilla-Gonzalez
V.,
Therapeutic potential of mesenchymal stem cells for cancer therapy. Frontiers in Bioengineering and Biotechnology.
2020;
8
:
43
.
View Article Google Scholar -
Xuan
X.,
Tian
C.,
Zhao
M.,
Sun
Y.,
Huang
C.,
Mesenchymal stem cells in cancer progression and anticancer therapeutic resistance. Cancer Cell International.
2021;
21
(1)
:
595
.
View Article Google Scholar -
He
N.,
Kong
Y.,
Lei
X.,
Liu
Y.,
Wang
J.,
Xu
C.,
MSCs inhibit tumor progression and enhance radiosensitivity of breast cancer cells by down-regulating Stat3 signaling pathway. Cell Death {&}amp; Disease.
2018;
9
(10)
:
1026
.
View Article Google Scholar -
Eggenhofer
E.,
Luk
F.,
Dahlke
M.H.,
Hoogduijn
M.J.,
The life and fate of mesenchymal stem cells. Frontiers in Immunology.
2014;
5
:
148
.
View Article Google Scholar -
Jumat
N.R.,
Yunus
M.A.,
Yahaya
B.H.,
Mohamed
R.,
Reprogramming macrophages toward M1-like phenotypes in the breast cancer microenvironment using mesenchymal stromal/stem cells: A review. Biomedical Research and Therapy.
2022;
9
(12)
:
5418-36
.
View Article Google Scholar -
Carty
F.,
Mahon
B.P.,
English
K.,
The influence of macrophages on mesenchymal stromal cell therapy: passive or aggressive agents?. Clinical and Experimental Immunology.
2017;
188
(1)
:
1-11
.
View Article Google Scholar -
Vasandan
A.B.,
Jahnavi
S.,
Shashank
C.,
Prasad
P.,
Kumar
A.,
Prasanna
S.J.,
Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Scientific Reports.
2016;
6
(1)
:
38308
.
View Article Google Scholar -
Rabani
R.,
Volchuk
A.,
Jerkic
M.,
Ormesher
L.,
Garces-Ramirez
L.,
Canton
J.,
Mesenchymal stem cells enhance NOX2-dependent reactive oxygen species production and bacterial killing in macrophages during sepsis. The European Respiratory Journal.
2018;
51
(4)
:
1702021
.
View Article PubMed Google Scholar -
Ullah
I.,
Subbarao
R.B.,
Rho
G.J.,
Human mesenchymal stem cells - current trends and future prospective. Bioscience Reports.
2015;
35
(2)
:
e00191
.
View Article Google Scholar -
Xi
J.,
Yan
X.,
Zhou
J.,
Yue
W.,
Pei
X.,
Mesenchymal stem cells in tissue repairing and regeneration: progress and future. Burns and Trauma.
2013;
1
(1)
:
13-20
.
View Article Google Scholar -
Lv
F.J.,
Tuan
R.S.,
Cheung
K.M.,
Leung
V.Y.,
Concise review: the surface markers and identity of human mesenchymal stem cells. Stem Cells (Dayton, Ohio).
2014;
32
(6)
:
1408-19
.
View Article Google Scholar -
Cleary
M.A.,
Narcisi
R.,
Focke
K.,
van der Linden
R.,
Brama
P.A.,
van Osch
G.J.,
Expression of CD105 on expanded mesenchymal stem cells does not predict their chondrogenic potential. Osteoarthritis and Cartilage.
2016;
24
(5)
:
868-72
.
View Article Google Scholar -
Dizaji Asl
K.,
Shafaei
H.,
Soleimani Rad
J.,
Nozad
H.O.,
Comparison of characteristics of human amniotic membrane and human adipose tissue derived mesenchymal stem cells. World Journal of Plastic Surgery.
2017;
6
:
33-9
.
-
Lee
H.J.,
Choi
B.H.,
Min
B.H.,
Park
S.R.,
Changes in surface markers of human mesenchymal stem cells during the chondrogenic differentiation and dedifferentiation processes in vitro. Arthritis and Rheumatism.
2009;
60
(8)
:
2325-32
.
View Article Google Scholar -
Mantovani
A.,
MSCs, macrophages, and cancer: a dangerous ménage-à-trois. Cell Stem Cell.
2012;
11
(6)
:
730-2
.
View Article Google Scholar -
Ren
G.,
Zhao
X.,
Wang
Y.,
Zhang
X.,
Chen
X.,
Xu
C.,
CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell.
2012;
11
(6)
:
812-24
.
View Article Google Scholar -
Parisi
L.,
Gini
E.,
Baci
D.,
Tremolati
M.,
Fanuli
M.,
Bassani
B.,
Macrophage polarization in chronic inflammatory diseases: killers or builders?. Journal of Immunology Research.
2018;
2018
:
8917804
.
View Article Google Scholar -
Peshkova
M.,
Korneev
A.,
Suleimanov
S.,
Vlasova
I.I.,
Svistunov
A.,
Kosheleva
N.,
MSCs' conditioned media cytokine and growth factor profiles and their impact on macrophage polarization. Stem Cell Research & Therapy.
2023;
14
(1)
:
142
.
View Article Google Scholar -
Huang
S.C.,
Smith
A.M.,
Everts
B.,
Colonna
M.,
Pearce
E.L.,
Schilling
J.D.,
Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis Is essential for macrophage alternative activation. Immunity.
2016;
45
(4)
:
817-30
.
View Article Google Scholar -
Prasanna
S.J.,
Gopalakrishnan
D.,
Shankar
S.R.,
Vasandan
A.B.,
Pro-inflammatory cytokines, IFNgamma and TNFalpha, influence immune properties of human bone marrow and Wharton jelly mesenchymal stem cells differentially. PLoS One.
2010;
5
(2)
:
e9016
.
View Article Google Scholar -
Ißleib
C.,
Kurz
S.,
Scholl
S.,
Amberg
B.,
Spohn
J.,
Plasticity of proinflammatory macrophages depends on their polarization stage during human MSC immunomodulation - an in vitro study using THP-1 and human primary macrophages. Immuno.
2021;
1
(4)
:
518-28
.
View Article Google Scholar -
Hill
B.S.,
Pelagalli
A.,
Passaro
N.,
Zannetti
A.,
Tumor-educated mesenchymal stem cells promote pro-metastatic phenotype. Oncotarget.
2017;
8
(42)
:
73296-73311
.
View Article Google Scholar -
Engström
A.,
Erlandsson
A.,
Delbro
D.,
Wijkander
J.,
Conditioned media from macrophages of M1, but not M2 phenotype, inhibit the proliferation of the colon cancer cell lines HT-29 and CACO-2. International Journal of Oncology.
2014;
44
(2)
:
385-92
.
View Article Google Scholar -
Song
W.,
Thakor
P.,
Vesey
D.A.,
Gobe
G.C.,
Morais
C.,
Conditioned medium from stimulated macrophages inhibits growth but induces an inflammatory phenotype in breast cancer cells. Biomedicine and Pharmacotherapy.
2018;
106
:
247-54
.
View Article Google Scholar -
Bagheri
R.,
Bitazar
R.,
Talebi
S.,
Alaeddini
M.,
Etemad-Moghadam
S.,
Eini
L.,
Conditioned media derived from mesenchymal stem cells induces apoptosis and decreases cell viability and proliferation in squamous carcinoma cell lines. Gene.
2021;
782
:
145542
.
View Article Google Scholar -
Widowati
W.,
Murti
H.,
Widyastuti
H.,
Laksmitawati
D.R.,
Rizal
R.,
Widya Kusuma
H.S.,
Decreased inhibition of proliferation and induction of apoptosis in breast cancer cell lines (T47D and MCF7) from treatment with conditioned medium derived from hypoxia-treated Wharton's Jelly MSCs compared with normoxia-treated MSCs. International Journal of Hematology-Oncology and Stem Cell Research.
2021;
15
:
77-89
.
View Article Google Scholar -
Khalil
C.,
Moussa
M.,
Azar
A.,
Tawk
J.,
Habbouche
J.,
Salameh
R.,
Anti-proliferative effects of mesenchymal stem cells (MSCs) derived from multiple sources on ovarian cancer cell lines: an in-vitro experimental study. Journal of Ovarian Research.
2019;
12
(1)
:
70
.
View Article Google Scholar -
Vis
M.A.,
Ito
K.,
Hofmann
S.,
Impact of culture medium on cellular interactions in in vitro co-culture systems. Frontiers in Bioengineering and Biotechnology.
2020;
8
:
911
.
View Article Google Scholar -
Rachakatla
R.S.,
Pyle
M.M.,
Ayuzawa
R.,
Edwards
S.M.,
Marini
F.C.,
Weiss
M.L.,
Combination treatment of human umbilical cord matrix stem cell-based interferon-beta gene therapy and 5-fluorouracil significantly reduces growth of metastatic human breast cancer in SCID mouse lungs. Cancer Investigation.
2008;
26
(7)
:
662-70
.
View Article Google Scholar -
Kalluri
R.,
LeBleu
V.S.,
The biology, function, and biomedical applications of exosomes. Science.
2020;
367
(6478)
:
eaau6977
.
View Article Google Scholar -
Keerthikumar
S.,
Chisanga
D.,
Ariyaratne
D.,
Al Saffar
H.,
Anand
S.,
Zhao
K.,
ExoCarta: A web-based compendium of exosomal cargo. Journal of Molecular Biology.
2016;
428
(4)
:
688-92
.
View Article Google Scholar -
Vakhshiteh
F.,
Atyabi
F.,
Ostad
S.N.,
Mesenchymal stem cell exosomes: a two-edged sword in cancer therapy. International Journal of Nanomedicine.
2019;
14
:
2847-59
.
View Article Google Scholar -
Lee
J.K.,
Park
S.R.,
Jung
B.K.,
Jeon
Y.K.,
Lee
Y.S.,
Kim
M.K.,
Exosomes derived from mesenchymal stem cells suppress angiogenesis by down-regulating VEGF expression in breast cancer cells. PLoS One.
2013;
8
(12)
:
e84256
.
View Article Google Scholar -
Zhou
W.,
Zhou
Y.,
Chen
X.,
Ning
T.,
Chen
H.,
Guo
Q.,
Pancreatic cancer-targeting exosomes for enhancing immunotherapy and reprogramming tumor microenvironment. Biomaterials.
2021;
268
:
120546
.
View Article Google Scholar -
Komohara
Y.,
Horlad
H.,
Ohnishi
K.,
Fujiwara
Y.,
Bai
B.,
Nakagawa
T.,
Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Science.
2012;
103
(12)
:
2165-72
.
View Article Google Scholar -
Zheng
Y.,
Cai
Z.,
Wang
S.,
Zhang
X.,
Qian
J.,
Hong
S.,
Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood.
2009;
114
(17)
:
3625-8
.
View Article Google Scholar -
Bogdanowicz
D.R.,
Lu
H.H.,
Multifunction co-culture model for evaluating cell-cell interactions. Methods in Molecular Biology (Clifton, N.J.).
2014;
1202
:
29-36
.
View Article Google Scholar -
Komohara
Y.,
Hasita
H.,
Ohnishi
K.,
Fujiwara
Y.,
Suzu
S.,
Eto
M.,
Macrophage infiltration and its prognostic relevance in clear cell renal cell carcinoma. Cancer Science.
2011;
102
(7)
:
1424-31
.
View Article Google Scholar -
Takaishi
K.,
Komohara
Y.,
Tashiro
H.,
Ohtake
H.,
Nakagawa
T.,
Katabuchi
H.,
Involvement of M2-polarised macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Science.
2010;
101
(10)
:
2128-36
.
View Article Google Scholar -
Yu
H.,
Kortylewski
M.,
Pardoll
D.,
Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nature Reviews. Immunology.
2007;
7
(1)
:
41-51
.
View Article Google Scholar -
Hinz
N.,
Jücker
M.,
Distinct functions of AKT isoforms in breast cancer: a comprehensive review. Cell Communication and Signaling.
2019;
17
(1)
:
154
.
View Article Google Scholar -
Choi
E.,
Kim
E.,
Kim
J.H.,
Yoon
K.,
Kim
S.,
Lee
J.,
AKT1-targeted proapoptotic activity of compound K in human breast cancer cells. Journal of Ginseng Research.
2019;
43
(4)
:
692-8
.
View Article Google Scholar -
deGraffenried
L.A.,
Friedrichs
W.E.,
Russell
D.H.,
Donzis
E.J.,
Middleton
A.K.,
Silva
J.M.,
Inhibition of mTOR activity restores tamoxifen response in breast cancer cells with aberrant Akt activity. Clinical Cancer Research.
2004;
10
(23)
:
8059-67
.
View Article Google Scholar -
Arranz
A.,
Doxaki
C.,
Vergadi
E.,
Martinez de la Torre
Y.,
Vaporidi
K.,
Lagoudaki
E.D.,
Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proceedings of the National Academy of Sciences of the United States of America.
2012;
109
(24)
:
9517-22
.
View Article Google Scholar -
Hu
B.,
Trinh
K.,
Figueira
W.F.,
Price
P.A.,
Isolation and sequence of a novel human chondrocyte protein related to mammalian members of the chitinase protein family. The Journal of Biological Chemistry.
1996;
271
(32)
:
19415-20
.
View Article Google Scholar -
Tsuruha
J.I.,
Masuko-Hongo
K.,
Kato
T.,
Sakata
M.,
Nakamura
H.,
Sekine
T.,
Autoimmunity against YKL-39, a human cartilage derived protein, in patients with osteoarthritis. The Journal of Rheumatology.
2002;
29
:
1459-66
.
-
Kavsan
V.,
Dmitrenko
V.,
Boyko
O.,
Filonenko
V.,
Avdeev
S.,
Areshkov
P.,
Overexpression of YKL-39 gene in glial brain tumors. Scholarly Research Exchange.
2008;
2008
:
1-8
.
View Article Google Scholar -
Liu
T.,
Larionova
I.,
Litviakov
N.,
Riabov
V.,
Zavyalova
M.,
Tsyganov
M.,
Tumor-associated macrophages in human breast cancer produce new monocyte attracting and pro-angiogenic factor YKL-39 indicative for increased metastasis after neoadjuvant chemotherapy. OncoImmunology.
2018;
7
(6)
:
e1436922
.
View Article Google Scholar -
Jensen
C.,
Teng
Y.,
Is It Time to Start Transitioning From 2D to 3D Cell Culture?. Frontiers in molecular biosciences.
2020;
7
:
33
.
View Article Google Scholar -
Jubelin
C.,
Muñoz-Garcia
J.,
Griscom
L.,
Three-dimensional in vitro culture models in oncology research. Cell & Bioscience.
2022;
12
(1)
:
155
.
View Article Google Scholar
Comments
Article Details
Volume & Issue : Vol 11 No 9 (2024)
Page No.: 6737-6752
Published on: 2024-09-30
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 2494 times
- PDF downloaded - 741 times
- XML downloaded - 127 times
