

Copyrights: Hafiz Muhammad Rehman, Memoona Naz, Ayesha Ghulam Ghous, Mahwash Malik, Sidra Ahmad, Hamid Bashir, 2025. License: This work is licensed under a Creative Commons Attribution 4.0 International License.
Abstract
Introduction: Synthetic fusion proteins represent a cutting-edge approach in biotechnology and pharmaceutical research, enabling the strategic combination of multiple protein domains to design novel complexes with enhanced properties and functionalities. By fusing distinct proteins, the unique attributes of each component can be synergistically exploited, leading to improved bioactivities or the emergence of entirely new functions. This study aimed to computationally construct a fusion protein that contained the killing properties of temporin 1CEa and the targeting action of IL-24, joined via a rigid linker, and its anti-tumor potential was analyzed using different bioinformatics tools.
Methods: After sequence retrieval, the 3D model of the temporin 1CEa-IL-24 fusion protein was constructed using the AlphaFold 2 online server. The resulting structure underwent rigorous refinement, quality assessment, and validation processes. Physiochemical properties were evaluated using ProtParam, and the prepared structure was subjected to docking on ClusPro and simulation on VMD and NAMD.
Results: Assessment through ERRAT score and Ramachandran plot analysis confirmed the good quality, refinement, and validation of the 3D structure of the temporin 1CEa-IL-24 fusion protein. Docking results revealed 17 hydrogen bonds and 4 salt bridges between the fusion protein and its receptor, findings supported and validated through simulation studies predicting a stable docked complex over a 100 ns period.
Conclusion: The expression of the temporin 1CEa-IL-24 fusion gene in a suitable expression host could lead to in vitro production and subsequently validate its therapeutic potential.
Introduction
Globally, cancer is a major cause of death following cardiovascular disorders. The term 'cancer' was derived from the Greek word "karkinos," which refers to malignant tumors. Cancer arises when there is uncontrolled proliferation in any organ of the body due to damage to normal cellular DNA1. The most prevalent types of cancer include lung, breast, liver, prostate, and colorectal cancers2. Among them, breast cancer is the most prevalent in females and the leading cause of death worldwide. Treatment for breast cancer entails surgery, chemotherapy, endocrine therapy, radiotherapy, and immunotherapy3, 4. Although these treatments have been proven to be effective, they cause severe harm that cannot be overlooked. The significant concern is that they are toxic and non-selective, damaging sensitive tissues, causing multidrug resistance, and tumor recurrence5, 6. Increased mortality rates due to cancer demand more advanced therapies that are selective, specific, and cost-effective, with fewer side effects7. Fusion proteins, effective in killing cancer cells, are therapeutic agents in which two or more proteins are fused to perform a targeted action that is more specific and selective8. Antimicrobial peptides have recently been proven to be efficient against breast cancer cells. This class has numerous advantages because it is selectively cytotoxic, may overcome multi-drug resistance, and can be employed in combination therapy9. The antimicrobial peptide temporin 1CEa precursor is obtained from the skin secretions of a Chinese frog, Rana chensinensis, and contains 17 amino acids. It is cytotoxic to breast cancer cells10. According to previous studies, it has been established that temporin 1CEa induces heightened permeability in MCF-7 cells upon exposure. This antimicrobial peptide appears to exert its impact by disrupting the integrity of the plasma membrane. As the concentration of temporin 1CEa increases, a consequential breakdown of the plasma membrane occurs, resulting in the loss of essential cytoplasmic elements. The observed increase in permeability and subsequent membrane disruption underscores the potential cytotoxic effects of temporin 1CEa on MCF-7 cells, shedding light on its mechanism of action and suggesting its potential utility in therapeutic applications targeting cancer cells with a focus on membrane destabilization9.
Cytokines, glycoproteins under 30 kDa, act by binding to receptors on cell membranes. They exert anti-tumor effects through pro-apoptotic actions or by stimulating immune responses against cancer cells11. This dual mechanism contributes to their role in inhibiting tumor cell growth and promoting a robust immune defense against malignancies12. The cytokine family of IL-10 includes IL-24, which is produced by immune cells of the body. IL-24 causes cell killing when it binds to receptors (IL-20R1/IL-20R2 and IL-22R1/IL-20R2) present on the surface of breast cancer cells and triggers the Janus kinase (JAK) and activator of transcription (STAT) signal transduction pathway13.
To design a novel targeted anticancer medicine, we aimed at producing a theoretical chimeric protein, temporin 1CEa-IL-24, to leverage its targeted delivery and specific action on breast cancer cells. To successfully create a chimeric protein, we need a linker sequence to join both parts of fusion proteins2. Linkers are classified into many types based on their properties; here we will focus only on rigid linkers for our protein structures to maintain a fixed distance between our domains so that they can perform independent functions14. The fusion of the lytic peptide temporin 1CEa with the targeting protein IL-24 to develop a recombinant protein may have the ability to kill breast cancer cells more effectively. Conducting computational studies prior to in vitro analyses not only offers predictive insights into expected results but also mitigates hazards and risks associated with laboratory-based testing15.
This study aims to computationally fuse the temporin 1CEa lytic peptide and the IL-24 targeting peptide through a rigid linker to exploit potential synergistic effects against breast cancer cell receptors, enhancing their anticancer efficacy. The newly designed fusion peptide (temporin 1CEa-IL-24) underwent rigorous quality assessment and validation before being docked to the heterodimer receptor (IL22RA-IL20RB) present in breast cancer cells. The docked complex was systematically analyzed for interactions, validation, and stability through simulations, with in silico expression predictions providing additional insights. Such computational approaches not only establish a foundation for future in vitro experiments but also offer resource-efficient alternatives, minimizing the need for laboratory animals and providing crucial insights that aid researchers in prioritizing and designing more targeted and successful experiments.
Methods
Construct Design and Sequence Analysis
The FASTA format of IL-24 [https://www.uniprot.org/uniprotkb/Q13007/entry](https://www.uniprot.org/uniprotkb/Q13007/entry) (Accession # Q13007) was retrieved from the UniProt data repository, and temporin 1CEa [https://www.ncbi.nlm.nih.gov/protein/ACF21594.1?report=fasta](https://www.ncbi.nlm.nih.gov/protein/ACF21594.1?report=fasta) (Accession # EU624139.1) was obtained from the NCBI database. To construct a fusion protein, temporin 1CEa was linked to IL-24 through a previously reported rigid-type linker (AEAAAKEAAAKA)16. The 3D (three-dimensional) structure of the IL-24 receptor (IL-22RA and IL-20RB, PDB ID: 6DF3) was retrieved in PDB format from the Protein Data Bank [https://www.rcsb.org/structure/6df3](https://www.rcsb.org/structure/6df3). The signal peptide (1–56 amino acids) was removed from the N-terminal of IL-24 to yield a mature peptide17.
Secondary Structure Prediction
The secondary structure of the chimeric protein was predicted using the GOR IV online server [http://gor.bb.iastate.edu/](http://gor.bb.iastate.edu/). Functional characteristics such as alpha and beta regions, regions lacking normal structure, coiled-coil domains, disulfide bridge locations, solvent-accessible surface area, and low-complexity sections were systematically evaluated. This comprehensive analysis offers insights into the protein's structural features and potential functional roles18, 19.
Tertiary Structure Prediction and Structure Refinement
The 3D structure of the chimeric protein was constructed using the AlphaFold 2 online server [https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb](https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb), developed by DeepMind, which employs advanced deep learning techniques. AlphaFold 2's neural network architecture predicts distances between amino acid pairs in the protein sequence, requiring only the FASTA sequence as input to generate 3D models. The resultant model underwent further refinement through the Galaxy Refine online server [https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE](https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE). This refinement process, crucial for minimizing protein energy and optimizing local and global structural features, enhanced the quality and accuracy of the protein structures. It specifically improved side-chain conformations, bond angles, and stereochemistry, preparing the structures for subsequent molecular docking studies20, 21.
Quality Assessment and Validation of Refined Protein Structure
For quality assessment and validation of the structure, the SAVES v6.0 [https://saves.mbi.ucla.edu/](https://saves.mbi.ucla.edu/) toolkit was used, which contains ERRAT and PROCHECK programs. ERRAT evaluated non-bonded interactions between various kinds of atoms and demonstrated error function compared to highly refined structures. A Ramachandran plot from PROCHECK was used to validate protein stereochemical properties and to check the quantity of residues in favored, allowed, and outer regions22. Additionally, ProSA (protein structure analysis), an online tool used for the prediction of potential errors in 3D protein models when provided in PDB format23, and QMEAN (qualitative model energy analysis) on ExPASy were employed to assess protein structure quality, providing a single score for models, aiding in validation and selection for accurate three-dimensional representations24.
Physiochemical Properties and Solubility Prediction of Fusion Proteins
The physiochemical properties of the fusion peptide were assessed using the ProtParam tool [https://web.expasy.org/protparam/protparam-doc.html](https://web.expasy.org/protparam/protparam-doc.html), accessible through the ExPASy server. The tool analyzed the peptide's primary sequence, providing essential intrinsic properties such as the instability index, estimated half-life, extinction coefficient, atomic composition, molecular weight, theoretical isoelectric point (pI), aliphatic index, amino acid composition, and the grand average of hydropathicity25. These parameters contribute valuable insights into the peptide's functional characteristics and behavior in diverse environments. Additionally, the solubility of the fusion protein was predicted using the Protein-Sol online server [https://protein-sol.manchester.ac.uk/](https://protein-sol.manchester.ac.uk/), which employs a dataset comparison approach to estimate solubility based on the input FASTA protein sequence26.
Toxicity, Antigenicity, and Allergenicity
In evaluating the fusion protein, a comprehensive analysis of its potential toxicity, antigenicity, and allergenicity was conducted using different bioinformatics tools. To assess the protein's antigenic nature, the VaxiJen server [http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html](http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) was employed, which relies on the computation of antigenic potential through the analysis of the protein's physical and chemical properties27. Allergenicity prediction was carried out using Allertop v2.0 [http://www.ddg-pharmfac.net/AllerTOP](http://www.ddg-pharmfac.net/AllerTOP), a tool specifically designed for assessing the likelihood of protein-inducing allergic responses28. ToxinPred [https://webs.iiitd.edu.in/raghava/toxinpred/prot_submitfreq_S.php?ran=63153](https://webs.iiitd.edu.in/raghava/toxinpred/prot_submitfreq_S.php?ran=63153), on the other hand, was utilized for the prediction of potential toxicity of the protein29. These analyses provide valuable insights into the safety and immune-related characteristics of the fusion protein with crucial information for potential applications in therapeutic or diagnostic contexts.
Molecular Docking Analysis
Molecular docking is an advanced computational method employed in drug discovery and structural biology to predict the preferred binding mode and orientation of one molecule (the ligand) when bound to another molecule (the receptor or target) for stable complex formation30. To elucidate the interactions and binding orientation between the temporin 1CEa-IL-24 fusion protein and its heterodimeric cognate receptor (IL22R1-IL21R2), a molecular docking study was conducted using the ClusPro 2.0 online server [https://cluspro.bu.edu/login.php?redir=/queue.php](https://cluspro.bu.edu/login.php?redir=/queue.php)31. The crystalline structure of the heterodimer receptor was retrieved in PDB format (PDB ID: 6DF3) for the docking analysis. Subsequently, using the PyMOL graphic system, the downloaded receptor structure underwent purification by removing attached IL24, ligands, and water molecules. The 3D structures of both the fusion protein and the receptor were submitted to the ClusPro 2.0 server with default settings. Finally, the MM/GBSA tool was employed to predict binding affinity and assess pre-energy residue contributions in the formation of the protein-protein complex [http://cadd.zju.edu.cn/hawkdock/](http://cadd.zju.edu.cn/hawkdock/)32.
Interaction Studies
Following the molecular docking process, the resulting docked complex of the temporin 1CEa-IL-24 fusion protein with its cognate receptor was subjected to interaction and affinity analysis using four distinct tools: PyMOL, PDBsum [http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/](http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/)33, PDBePISA [https://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver](https://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver)34, and HawDock online server [http://cadd.zju.edu.cn/hawkdock/](http://cadd.zju.edu.cn/hawkdock/)32. These servers provided various aspects of the protein-protein interaction, including the identification of interacting interfaces, pores, assessment of binding affinity, salt bridges, solvation energy in kcal/mol, hydrogen bonds, and tunnels present in the docked complex.
Molecular Dynamics Simulation
The fusion protein underwent molecular dynamics (MD) simulation using the Visual Molecular Dynamics (VMD) software35 and the NAMD (Nanoscale Molecular Dynamics) simulation engine36. The input files for the simulation, including the addition of missing hydrogens achieved through the LeaP program in Ambertools 2137. A solvation box with a 10 Å buffer, populated with TIP3P water molecules, was employed to solvate the complex. To maintain system neutrality, counter ions, Na+ and Cl-, were strategically added. To alleviate clashes within the system, the ff14SB force field38 was applied for energy minimization. The solvated systems underwent three temperature equilibrations at 300, 200, and 250K to stabilize the environment. Subsequently, a 100 ns MD simulation ensued, saving MD trajectories at 2 ps intervals. Finally, the R package39 was utilized to analyze the trajectory data, providing valuable insights into the dynamic behavior, stability, and conformational changes of the fusion protein during the simulation period.

Results
Fusion Construct
A fusion protein of 184 amino acids was constructed by joining the N-terminal of temporin 1CEa to the C-terminal of a rigid linker, and the N-terminal of the linker was further attached to the C-terminal of the IL-24 peptide. A rigid linker (AEAAAKEAAAKA) was used to ensure the integrity of both functional domains so that it could prevent disulfide bond formation within protein molecules and provide stability to the structure (Figure 1).
Secondary Structure Prediction
The GOR IV online server predicted the secondary structure based on alpha, beta coils, extended strands, beta turns, and beta bridges. According to the results obtained, more than 50% of the fusion protein model contained alpha sheets composed of hydrophobic amino acids, forming the backbone of a protein structure (Figure 1). The predicted secondary structure of the protein reveals a predominant alpha-helical conformation, constituting 54.89% of the protein's structure. Additionally, extended strands make up 15.22%, while a significant portion is predicted to be in a random coil configuration, accounting for 29.89%.
3D Structure Prediction of Fusion Protein
AlphaFold 2, developed by DeepMind and accessible through Google's Colab platform, is an innovative AI program designed for predicting the 3D structure of proteins. Leveraging a combination of chemical bond angles and amino acid sequence features, the tool generated ten structural models as output. The output file from AlphaFold 2 provided five distinct 3D models of the protein structure, ranked based on local model quality. These rankings reflect the reliability and accuracy of predictions at the individual structural level.


Quality Check and Validation of Temporin 1CEa-IL24 Fusion Protein
The Temporin 1CEa-IL24 fusion construct was validated by Ramachandran Plot (RC plot) analysis. An RC plot is a two-dimensional plot of phi-psi angles; these two angles are theoretically crucial and segregate protein structure into allowed and disallowed regions. This phi-psi space provides a base to assess the reliability and integrity of protein structure40, 41. The selected model showed 97.7% residues in the most favored region, 2.3% residues in the additionally allowed region, and no residues in the generously allowed region (Figure 2 A). For quality assessment, the refined 3D model of the chimeric protein was uploaded to respective online servers of ERRAT, ProSA-web, and ExPASy. ERRAT is a crystallography validation program assessing the overall quality of protein structures. It evaluates non-bonded atomic interactions, with higher scores indicating greater structure quality. The 97.72% ERRAT score of the temporin 1CEa-IL24 fusion protein (Figure 2 B) suggests high structural quality, surpassing the generally accepted range (>50) for a well-constructed model42. The evaluation of the structural quality of the temporin 1CEa-IL24 fusion protein model was conducted through ProSA-web analysis and QMEAN scoring. ProSA-web, a widely utilized tool for assessing 3D protein structure models, placed the fusion protein within the range of native conformations, as evidenced by a Z-score of -5.54 (Figure 3 A). The largely negative residue energy, especially in the C-terminal domain depicted in Figure 3B, further indicated structural stability, with an energy distribution below the zero baseline. This observation aligns with the expected native states of the protein. The QMEAN scoring function, evaluating both global and per-residue metrics, provided additional insights into model quality. A QMEAN score close to zero signifies a high-quality fit between the model's structure and the experimental reference, indicating a reliable representation of the protein's conformation and provides a quantitative measure of the overall accuracy and alignment of the model with experimental structural data43. The QMEAN4 score of 0.09, illustrated in Figure 2C, signified a high-quality fit between the model's structure and the experimental reference, affirming the reliability of the temporin 1CEa-IL24 fusion protein model. A MolProbity score of 1.2 indicates a high-quality structure with good stereochemistry, minimal steric clashes, and well-optimized backbone and side-chain conformations. This score is comparable to high-resolution crystal structures, suggesting that the fusion protein has reliable geometry for molecular modeling, docking, and MD simulations. The low clash score and high Ramachandran and rotamer quality further validate its accuracy. This quantitative measure not only indicates overall accuracy but also highlights the alignment of the model with experimental structural data, reinforcing the credibility of the proposed protein conformation.
| Physiochemical Properties | Values |
|---|---|
| Number of amino acids | 184 |
| Theoretical pI | 9.06 |
| Molecular weight | 21148.48 |
| Instability index | 37.16 |
| Aliphatic index | 95.98 |
| Total positively charged amino acids (Arg + Lys) | 23 |
| Total negatively charged amino acids (Asp + Glu) | 19 |
| Grand average of hydropathicity (GRAVY) | -0.08 |
| Predicted half-life | 2 mins ( E. coli, in vivo ) |
| Coefficient of extinction (in M⁻¹ cm⁻¹ at 280 nm) | 22585 |
Physicochemical Properties of Fusion Construct
The ProtParam-predicted physicochemical properties of the temporin 1CEa-IL24 fusion protein in Table 1, consisting of 184 amino acids, reveal characteristics associated with stability and functional relevance. With a theoretical isoelectric point of 9.06, the protein tends to be positively charged under physiological conditions. The molecular weight of 21148.48 Da provides insight into its mass, aiding in characterization. The instability index of 37.16 suggests stability, while the aliphatic index of 95.98 indicates a favorable structural arrangement. The balance between positively23 and negatively19 charged amino acids contributes to the overall charge distribution. The slightly hydrophilic nature, as reflected in the Grand Average of Hydropathicity (GRAVY) of -0.08, suggests potential solubility characteristics. However, the predicted short half-life of 2 minutes in E. coli (in vivo) may impact practical applications. The coefficient of extinction at 280 nm (22585 M-1 cm-1) provides useful information for experimental analysis and could be correlated during in vitro studies. Overall, these physicochemical properties present a picture of a protein with intriguing features, warranting further investigation and experimental validation for a comprehensive understanding of its behavior and potential therapeutic applications.
Toxicity, antigenicity, and allergenicity
The results obtained from ToxinPred, Vaxigen, and Allertop v. 2.0 online servers collectively indicate a favorable profile for the chimeric protein, suggesting it to be non-toxic, non-allergenic, and non-antigenic. These findings are crucial for assessing the safety of the protein in various applications, such as in the development of therapeutics. The protective antigen prediction of 0.5502 falls slightly below the set threshold of 0.6, indicating a moderate level of confidence in its protective antigenic nature. While the protein may not exhibit strong antigenic properties, its overall non-toxic and non-allergenic characteristics are promising for potential biomedical applications.
Docking Analysis
Molecular docking is a sophisticated bioinformatics modeling approach designed to elucidate the interactions between two or more molecules, ultimately resulting in the formation of stable adducts. This method relies on predicting the three-dimensional structure of complexes formed by ligands and target receptors, offering valuable insights into the binding properties of these molecules. Through the generation of diverse possible adduct structures, molecular docking employs a scoring function within specialized software to rank and group these structures44. The ClusPro docking web server, renowned for its robust methodology, ensures the precision of protein-protein binding affinity evaluations. It operates on the foundational principle of the Fast Fourier Transform Correlation method, employing a three-step process to compute and rank docked complexes. Initially, rigid docking is executed using PIPER, sampling billions of conformations to comprehensively explore potential binding orientations. Subsequently, the algorithm generates clusters consisting of one thousand lowest energy structures, employing RMSD (root mean square deviation) as a criterion. This step enables the prediction of the largest cluster, presenting the most probable docked complex. Finally, the selected docked model undergoes a crucial energy minimization step, enhancing stability by refining the molecular arrangement31. The docking of the temporin 1CEa-IL24 fusion protein with its heterodimer receptor was conducted using the ClusPro online server, yielding ten dock models each accompanied by weighted scores as mentioned in Table 2. Among these models, the best-selected balanced model (Figure 4) exhibited a center-weighted score of -829.0 and the lowest energy score of -926.0 kcal/mol. These scores strongly suggest the potential for effective interaction between the temporin 1CEa-IL24 fusion protein and the IL-24 heterodimer receptor, a critical aspect to consider in its candidacy for anti-cancer therapeutics. The MMGBSA binding affinity of -96.21 kcal/mol obtained from a molecular docking analysis signifies a highly favorable interaction between the ligand and the target protein. This negative value implies that the binding process is thermodynamically favorable, indicating an exothermic reaction wherein energy is released upon complex formation.
| Cluster | Members | Characteristic | Weighing Score |
| 00 | 43 | Center | -829.0 |
| Low Energy | -926.1 | ||
| 01 | 33 | Center | -741.7 |
| Low Energy | -847.8 | ||
| 02 | 32 | Center | -787.5 |
| Low Energy | -864.4 | ||
| 03 | 28 | Center | -746.9 |
| Low Energy | -794.4 | ||
| 04 | 28 | Center | -788.8 |
| Low Energy | -812.5 | ||
| 05 | 27 | Center | -763.7 |
| Low Energy | -932.7 | ||
| 06 | 26 | Center | -758.8 |
| Low Energy | -877.3 | ||
| 07 | 26 | Center | -734.0 |
| Low Energy | -847.1 | ||
| 08 | 22 | Center | -749.8 |
| Low Energy | -934.1 | ||
| 09 | 21 | Center | -745.9 |
| Low Energy | -871.0 | ||
| 10 | 21 | Center | -818.9 |
| Lowest Energy | -818.9 |



Interaction Studies
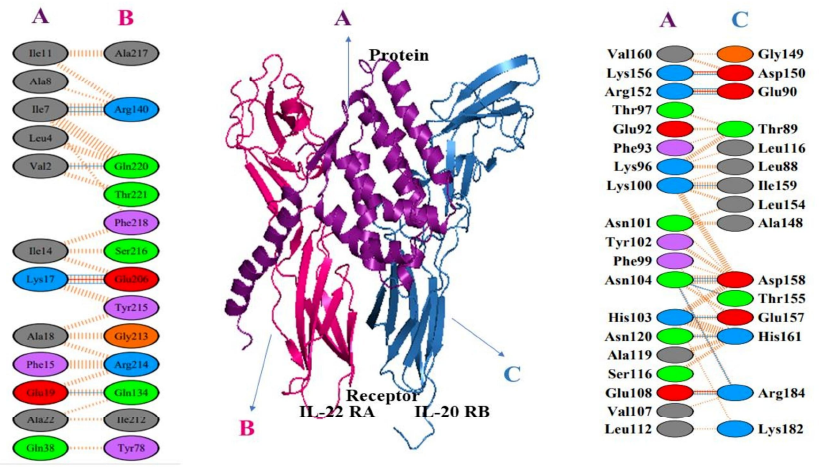
The molecular interactions and energy changes during docked complex formation were meticulously resolved using online servers of PDBsum and PDBePISA. The analysis unveiled a total of 21 interactions, comprising 4 salt bridges and 17 hydrogen bonds, without the presence of disulfide bonds between the temporin 1CEa-IL24 fusion protein and the heterodimer receptor subunits. Specifically, with the IL-22R1 subunit, a total of 7 bonds were predicted, consisting of 1 salt bridge and 6 hydrogen bonds. The interface residues between these chains extended over 568.5 Å, contributing to a solvation-free energy gain of -6.0 kcal/mol. Notable residues involved in hydrogen bond formation included Ile7, Val2, Lys17, and Glu19 from the fusion construct, as well as Arg140, Gln220, Glu206, and Gln134 from the IL-22R1 subunit. Similarly, with the IL-20R2 subunit, a total of 14 bonds were predicted, encompassing 3 salt bridges and 11 hydrogen bonds. The interface residues spanned 926.7 Å, with a solvation-free energy gain of -6.0 kcal/mol. Key residues in hydrogen bond formation included Asp150, Glu90, Ile159, Asp158, Glu157, His161, and Arg184 from IL-20R2, and Lys156, Arg152, Lys100, Asn104, His103, Asn120, Glu108 from the fusion construct. These detailed molecular insights shed light on the intricacies of the temporin 1CEa-IL24 fusion protein's binding interactions with its heterodimer receptor, offering valuable information for potential therapeutic applications, particularly in the context of breast cancer treatment. The visualization of interacting amino acids between the chimeric protein and its receptor using PyMOL software provided a detailed spatial understanding of the binding interface as illustrated in Figure 5 and Figure 6.
Molecular Dynamics Simulation
In the 100 ns molecular dynamics simulation of the chimeric protein complex, a comprehensive analysis was conducted to assess its stability and dynamics. The root mean square deviation (RMSD) of the backbone atoms served as a key metric to evaluate the structural stability of the complex throughout the simulation period. Notably, the complex demonstrated a rapid equilibration phase, stabilizing at around 10 ns with an RMSD value of approximately 4 Å. Following this equilibration, the RMSD exhibited deviations within the range of 4-8 Å, indicative of dynamic fluctuations, before ultimately settling into a more consistent stability range of 6-7 Å by 42 ns. This stability persisted until the conclusion of the simulation. The smooth trajectory of the RMSD, as illustrated in Figure 7A, suggests that the chimeric protein complex maintained structural integrity and stability throughout the entire 100 ns simulation, providing valuable insights into its behavior and suitability for further analyses or applications. The assessment of the chimeric protein complex's compactness involved a thorough analysis of its radius of gyration (Rg), providing crucial insights into its overall size and structural integrity during the 100 ns molecular dynamics simulation. At equilibrium, the Rg value stabilized at around 29.5 Å, indicating a compact and well-defined structure. Subsequently, a gradual increase in Rg was observed, reaching a maximum of 30 Å at 40 ns, suggesting a slight expansion of the overall size of the complex. However, the Rg values thereafter consistently maintained an average of 29.2 Å until the end of the simulation (Figure 7B). The minimal overall Rg difference of less than 0.8 Å throughout the simulation underscores the sustained compactness of the chimeric protein complex. This observation further supports the notion that the structural integrity of the complex was well-maintained, contributing to its stability and suggesting its suitability for potential functional and mechanistic studies in various biological contexts. To measure the flexibility of protein residues within the chimeric protein complex, root mean square fluctuations (RMSF) were calculated, offering insights into the dynamic behavior of individual residues throughout the 100 ns molecular dynamics simulation. Higher RMSF values signify greater flexibility, while lower values suggest rigidity, particularly within protein secondary structures. The RMSF analysis revealed distinct regions of flexibility, notably residues spanning from 20 to 40, 80 to 90, 250 to 270, and 390 to 410, displaying elevated RMSF values with a maximum reaching 15 Å. This increased flexibility is attributed to the presence of large loop regions in these specific parts of the protein (Figure 7 C). Conversely, the majority of protein residues remained relatively rigid, with RMSF values not exceeding 2 Å. The overall RMSF analysis underscores the stability of the protein complex, with specific regions of flexibility likely contributing to its dynamic behavior while maintaining a globally stable conformation throughout the simulation period. Collectively, these analyses affirm that the chimeric protein complex maintained an overall stable, compact, and structurally rigid conformation during the entire simulation, providing valuable insights into its dynamic behavior and structural integrity.

Discussion
Breast cancer, primarily affecting the epithelial tissue of the breast, predominantly affects women. The emergence of drug resistance poses a significant challenge in its treatment, underscoring the urgent need for novel therapeutic approaches. Peptides have shown promise as potential anti-cancer agents, offering a new avenue for drug discovery and treatment strategies against breast cancer45. Breast cancer treatment encompasses various modalities such as chemotherapy, molecular targeted therapy, endocrine therapy, radiotherapy, and surgery. Chemotherapy has emerged as a key treatment option for breast cancer across all stages, particularly as the disease often presents as systemic at diagnosis. By targeting residual tumor cells in the body, systemic chemotherapy can enhance the efficacy of surgery and improve cure rates. However, drug resistance remains a significant challenge during chemotherapy, highlighting the critical need for the development of novel anticancer drugs to enhance treatment outcomes for breast cancer46. Previously, interleukin-peptide fusion proteins have been assessed using computational methods, leading to their successful therapeutic potential prediction for in vitro expression and purification. Such as interleukin 24-BR2 peptide, showing significant activity against MCF-7 breast cancer cells47, interleukin 24-p28 peptide, interleukin 24-NRC peptide, NBD-IL24 peptide, TAT-interleukin 24-KDEL peptide, Melittin-IL24, and interleukin 24-RGD peptide have shown promising efficacy. Studies by Ghavimi et al. (2020) and Jahanian-Najafabadi et al. (2020) highlight the effectiveness of interleukin 24-p28 peptide against breast cancer16, 48 Similarly, Soleimani et al. (2016) demonstrated the activity of interleukin 24-NRC peptide against breast cancer cells49, Rehman et al. (2024) investigated NBD-IL24 peptide as a potential therapeutic agent for breast cancer treatment50. Additionally, Rehman et al. (2023) explored Melittin-IL2451, while Xiao et al. (2009) focused on interleukin 24-RGD peptide, both showing promising results in targeting breast cancer cells, including specific interactions with MCF-7 cancer cells52. This computational study has unveiled the promising potential of a novel fusion peptide, temporin 1CEa-IL24, designed to target breast cancer cell receptors. The meticulous structural and computational analyses performed in this study have consistently indicated that the three-dimensional structure of the temporin 1CEa-IL24 fusion protein adheres to high-quality standards, boasting commendable refinement and validation scores. The secondary structure prediction revealed a predominantly alpha-helical conformation, indicating a well-defined structure with a clear dominance of alpha helices. Interestingly, the absence of beta sheets, beta turns, and ambiguous states suggests a relatively well-defined secondary structure with a clear dominance of alpha helices. The high percentage of alpha helix content is indicative of a protein region characterized by a helical arrangement of the peptide backbone, often associated with stable and structured domains53. Through advanced AI-driven tools such as AlphaFold 2, the 3D structure prediction provided valuable insights into the protein's spatial arrangement and conformation, facilitating further refinement and validation processes. Quality assessment and validation analyses, including Ramachandran Plot analysis, ProSA-web evaluation, and QMEAN scoring, collectively affirmed the structural integrity and reliability of the proposed protein model. Physicochemical properties highlighted features associated with stability and functional relevance, while toxicity, antigenicity, and allergenicity assessments indicated a favorable safety profile.
To enhance the stability of the temporin 1CEa-IL24 fusion protein in E. coli, several strategies can be applied. Codon optimization can improve translational efficiency, while fusion tags like SUMO or MBP enhance solubility and stability. Co-expressing chaperones (DnaK/DnaJ) aids proper folding, and protease-resistant mutations or disulfide bond engineering can prevent degradation. Lowering expression temperature (16–25°C) and using protease-deficient E. coli strains (e.g., BL21(DE3) pLysS) further improve stability. For therapeutic use, PEGylation or glycosylation can extend half-life. These approaches could enhance protein expression and practical application54. ClusPro was employed to perform docking as the primary goal of molecular docking is to achieve a ligand-receptor complex with an optimized conformation, aiming for minimal binding free energy. By systematically exploring various ligand orientations and conformations within the binding site of the target receptor, molecular docking assists in identifying potential drug candidates and contributes to the rational design of novel therapeutic agents with enhanced binding affinity and stability55. Molecular docking analyses demonstrated the potential for effective interaction between the fusion protein and its target receptor, crucial for therapeutic considerations. Specifically, the center score reflects the highest structural energy within neighboring structures, providing insights into the stability and arrangement of the complex. On the other hand, the weighted score of the lowest energy highlights the most energetically favorable structure within its respective cluster. These docking results, with favorable energy scores, lay the foundation for further exploration of the temporin 1CEa-IL24 fusion protein as a promising candidate for targeted anti-cancer therapeutic applications. Additionally, a pronounced negative binding affinity suggests a robust and stable interaction, showcasing the strength of the ligand-protein binding. The result reflects the combined effects of various molecular forces, including hydrogen bonding, van der Waals forces, and electrostatic interactions. Overall, a MMGBSA binding affinity of -96.21 kcal/mol points towards a strong and energetically favorable binding between the ligand and the protein, emphasizing the potential biological significance of the observed interaction. The 95 hydrophobic interactions in complex indicate a highly stable and strong binding interface. These interactions minimize water exposure, enhancing molecular complementarity and structural integrity. Such extensive hydrophobic forces likely contribute to prolonged receptor binding, ensuring effective IL-24-mediated signaling for tumor suppression. Additionally, they may enhance the therapeutic potential of the fusion protein by preventing premature dissociation.
The stability of ligand binding within the binding pocket of a protein-protein complex was investigated through parameters such as Cα-RMSD and Cα-root-mean-square fluctuation (RMSF) values, alongside molecular dynamics (MD) simulations tracking ligand-receptor interactions over time. Analysis revealed a pattern where after an initial increase in RMSD, structural fluctuations reached a plateau, indicating a stable ligand binding state. This trend was consistent across various time points, suggesting a robust and sustained interaction between the ligand and its receptor. Specifically, examination of the fusion peptide structure within the heterodimer receptor junction envelope showed minimal fluctuations, indicating tight and stable binding, crucial for potential therapeutic applications. The observed stability and adaptation of the ligand to its binding site, coupled with the consistency in structural fluctuations across molecular species, support the prediction of potent anticancer activity for the fusion protein. The docking and simulation results have offered invaluable insights into the intricate interactions between the fusion protein and its receptor, showing the presence of multiple hydrogen bonds and salt bridges that contribute to the stability of the complex. Significantly, the predicted stability of the docked complex over an extended simulation period provides a solid foundation for guiding subsequent experimental investigations.
Future validation of the temporin 1CEa-IL24 fusion protein will involve in vitro expression in a suitable host (e.g., E. coli or HEK293), followed by purification and characterization using SDS-PAGE and Western blotting. Cytotoxicity assays on breast cancer cell lines (e.g., MCF-7, MDA-MB-231) will assess its tumor-killing efficiency, while receptor binding studies (e.g., ELISA or SPR) will confirm target specificity. Additionally, potential in vivo studies using xenograft mouse models could evaluate tumor-targeting efficiency, biodistribution, and therapeutic effects, providing experimental validation of the computational predictions.
CONCLUSION
In conclusion, the strategy of expressing the Temporin-1CEa-IL24 fusion gene in a suitable host presents a potential approach for further investigation in the context of breast cancer treatment. This computational study provides insights into the design and evaluation of targeted fusion proteins, highlighting their theoretical potential based on in silico analyses. While computational studies offer valuable preliminary data, experimental validation is essential to confirm their therapeutic applicability. Integrating computational and experimental approaches can aid in the rational development of novel cancer therapeutics.
Abbreviations
GRAVY: Grand Average of Hydropathicity, Jak: Janus Kinase, MM/GBSA: Molecular Mechanics/Generalized Born Surface Area, MD: Molecular Dynamics, NAMD: Nanoscale Molecular Dynamics, PIPER: Protein Interaction Prediction and Energy Refinement, ProSA: Protein Structure Analysis, QMEAN: Qualitative Model Energy Analysis, Rg: Radius of Gyration, RMSD: Root Mean Square Deviation, RMSF: Root Mean Square Fluctuations, STAT: Signal Transducer and Activator of Transcription, VMD: Visual Molecular Dynamics, pI: Isoelectric Point.
Acknowledgments
All authors acknowledge and thank the groups that have developed state-of-the-art computational tools and online servers for these analyses.
Author’s contributions
All authors contributed to the study conception and design. Data collection and analysis were performed by all authors. All authors read and approved the final manuscript.
Funding
The current project was funded by HEC Pakistan through NRPU 2021 (Project No. 16935) and the University of the Punjab, Lahore, Pakistan.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
-
Sudhakar
A.,
History of cancer, ancient and modern treatment methods. Journal of Cancer Science & Therapy.
2009;
1
(2)
:
1-4
.
View Article PubMed Google Scholar -
Ghavimi
R.,
Mohammadi
E.,
Akbari
V.,
Shafiee
F.,
In silico design of two novel fusion proteins, p28-IL-24 and p28-M4, targeted to breast cancer cells. Research in Pharmaceutical Sciences.
2020;
15
(2)
:
200-8
.
View Article PubMed Google Scholar -
Khalid
S.,
Rehman
H.M.,
Al-Qassab
Y.,
Ahmad
I.,
Fatima
T.,
Mubasher
M.M.,
Design and computational analysis of a novel Leptulipin-p28 fusion protein as a multitarget anticancer therapy in breast cancer. Toxicology Research.
2024;
13
(5)
:
tfae174
.
View Article PubMed Google Scholar -
Fatima
T.,
Mubasher
M.M.,
Rehman
H.M.,
Niyazi
S.,
Alanzi
A.R.,
Kalsoom
M.,
Computational modeling study of IL-15-NGR peptide fusion protein: a targeted therapeutics for hepatocellular carcinoma. AMB Express.
2024;
14
(1)
:
91
.
View Article PubMed Google Scholar -
Sain
R.K.,
Chouhan
R.,
Bagri
L.P.,
Bajpa
A.,
Strategies of targeting tumors and cancers. Journal of Cancer Research Updates.
2012;
1
(1)
:
129-52
.
-
Solanki
R.,
Saini
M.,
Mochi
J.,
Pappachan
A.,
Patel
S.,
Synthesis, characterization, in-silico and in-vitro anticancer studies of Plumbagin encapsulated albumin nanoparticles for breast cancer treatment. Journal of Drug Delivery Science and Technology.
2023;
84
:
104501
.
View Article Google Scholar -
Qureshi
S.,
Ahmed
N.,
Rehman
H.M.,
Amirzada
M.I.,
Saleem
F.,
Waheed
K.,
Investigation of therapeutic potential of the Il24-p20 fusion protein against breast cancer: an in-silico approach. In Silico Pharmacology.
2024;
12
(2)
:
84
.
View Article PubMed Google Scholar -
Rehman
H.M.,
Yousaf
N.,
Hina
S.M.,
Nadeem
T.,
Ansari
M.A.,
Chaudry
A.,
Design and computational analysis of a novel Azurin-BR2 chimeric protein against breast cancer. Toxicology Research.
2024;
13
(6)
:
tfae179
.
View Article PubMed Google Scholar -
Wang
C.,
Tian
L.L.,
Li
S.,
Li
H.B.,
Zhou
Y.,
Wang
H.,
Rapid cytotoxicity of antimicrobial peptide tempoprin-1CEa in breast cancer cells through membrane destruction and intracellular calcium mechanism. PLoS One.
2013;
8
(4)
:
e60462
.
View Article PubMed Google Scholar -
Wang
C.,
Li
H.B.,
Li
S.,
Tian
L.L.,
Shang
D.J.,
Antitumor effects and cell selectivity of temporin-1CEa, an antimicrobial peptide from the skin secretions of the Chinese brown frog (Rana chensinensis). Biochimie.
2012;
94
(2)
:
434-41
.
View Article PubMed Google Scholar -
Rehman
H. Muhammad,
Rehman
H.M.,
Naveed
M.,
Khan
M.T.,
Shabbir
M.A.,
Aslam
S.,
In Silico Investigation of a Chimeric IL24-LK6 Fusion Protein as a Potent Candidate Against Breast Cancer. Bioinformatics and Biology Insights.
2023;
17
:
11779322231182560
.
View Article PubMed Google Scholar -
Berraondo
P.,
Sanmamed
M.F.,
Ochoa
M.C.,
Etxeberria
I.,
Aznar
M.A.,
Pérez-Gracia
J.L.,
Cytokines in clinical cancer immunotherapy. British Journal of Cancer.
2019;
120
(1)
:
6-15
.
View Article PubMed Google Scholar -
Persaud
L.,
De Jesus
D.,
Brannigan
O.,
Richiez-Paredes
M.,
Huaman
J.,
Alvarado
G.,
Mechanism of action and applications of interleukin 24 in immunotherapy. International Journal of Molecular Sciences.
2016;
17
(6)
:
869
.
View Article PubMed Google Scholar -
Chen
X.,
Zaro
J.L.,
Shen
W.C.,
Fusion protein linkers: property, design and functionality. Advanced Drug Delivery Reviews.
2013;
65
(10)
:
1357-69
.
View Article PubMed Google Scholar -
Shahid
F.,
Iftikhar
I.,
Rehman
H.M.,
Javaid
S.,
Fatima
M.,
Rehman
I.,
Biosecurity and Biosafety concerns of Research and diagnostic Laboratory under International Guidelines. Advancements in Life Sciences.
2022;
9
(2)
:
151-6
.
View Article Google Scholar -
Ghavimi
R.,
Mohammadi
E.,
Akbari
V.,
Shafiee
F.,
Jahanian-Najafabadi
A.,
In silico design of two novel fusion proteins, p28-IL-24 and p28-M4, targeted to breast cancer cells. Research in Pharmaceutical Sciences.
2020;
15
(2)
:
200-8
.
View Article PubMed Google Scholar -
Ghosh
P.,
Patra
P.,
Mondal
N.,
Chini
D.S.,
Patra
B.C.,
Multi Epitopic Peptide Based Vaccine Development Targeting Immobilization Antigen of Ichthyophthirius multifiliis: A Computational Approach. International Journal of Peptide Research and Therapeutics.
2023;
29
(1)
:
11
.
View Article PubMed Google Scholar -
Kloczkowski
A.,
Ting
K.L.,
Jernigan
R.L.,
Garnier
J.,
Combining the GOR V algorithm with evolutionary information for protein secondary structure prediction from amino acid sequence. Proteins.
2002;
49
(2)
:
154-66
.
View Article PubMed Google Scholar -
Mohseni Moghadam
Z.,
Halabian
R.,
Sedighian
H.,
Behzadi
E.,
Amani
J.,
Imani Fooladi
A.A.,
Designing and analyzing the structure of DT-STXB fusion protein as an anti-tumor agent: an in silico approach. Iranian Journal of Pathology.
2019;
14
(4)
:
305-12
.
View Article PubMed Google Scholar -
Aslam
S.,
Qureshi
S.,
Bashir
H.,
In silico investigation of a novel anti EGFR Scfv–IL 24 fusion protein induces apoptosis in malignant cells. Journal of Molecular Modeling.
2023;
29
(9)
:
282
.
View Article Google Scholar -
Alshehri
A.A.,
Almutairi
A.M.,
Shafie
A.,
Alshehri
N.A.,
Almutairi
S.M.,
Anjum
F.,
Identification of potential inhibitors targeting DNA adenine methyltransferase of Klebsiella pneumoniae for antimicrobial resistance management: a structure-based molecular docking study. Advancements in Life Sciences.
2024;
10
(4)
:
604-8
.
View Article Google Scholar -
Awais
H.,
Zahid
A.,
Afzaal
A.,
Mannan
T.,
Habib
H.,
Coding Genome Sequence and Protein Sequence Analysis of Dengue Strains: In Silico Correlation. Advancements in Life Sciences.
2023;
10
(1)
:
48-53
.
View Article Google Scholar -
Wiederstein
M.,
Sippl
M.J.,
ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research.
2007;
35
(Web Server issue)
:
W407-10
.
View Article PubMed Google Scholar -
Saif
R.,
Ashfaq
K.,
Ali
G.,
Iftekhar
A.,
Zia
S.,
Yousaf
M.Z.,
Computational prediction of Cassia angustifolia compounds as a potential drug agents against main protease of SARS-nCov2. Advancements in Life Sciences.
2022;
9
(1)
:
36-40
.
View Article Google Scholar -
Javaid
M.S.,
Kaul
H.,
Fazal
N.,
Yaqub
F.,
Naseer
N.,
Hanif
M.,
In silico analysis to reveal underlying trans differentiation mechanism of Mesenchymal Stem Cells into Osteocytes. Advancements in Life Sciences.
2021;
8
(4)
:
412-8
.
View Article Google Scholar -
Aslam
S.,
Rehman
H.M.,
Sarwar
M.Z.,
Ahmad
A.,
Ahmed
N.,
Amirzada
M.I.,
Computational Modeling, High-Level Soluble Expression and In Vitro Cytotoxicity Assessment of Recombinant Pseudomonas aeruginosa Azurin: A Promising Anti-Cancer Therapeutic Candidate. Pharmaceutics.
2023;
15
(7)
:
1825
.
View Article PubMed Google Scholar -
Doytchinova
I.A.,
Flower
D.R.,
VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics.
2007;
8
(1)
:
4
.
View Article PubMed Google Scholar -
I. Dimitrov,
D.R. Flower,
I. Doytchinova,
AllerTOP-a server for in silico prediction of allergens. BMC bioinformatics.
2013;
14
:
1-9
.
View Article Google Scholar -
Gupta
S.,
Kapoor
P.,
Chaudhary
K.,
Gautam
A.,
Kumar
R.,
Raghava
G.P.,
Consortium
undefined Open Source Drug Discovery,
In silico approach for predicting toxicity of peptides and proteins. PLoS One.
2013;
8
(9)
:
e73957
.
View Article PubMed Google Scholar -
Pérez
S.,
Tvaroška
I.,
Carbohydrate-protein interactions: molecular modeling insights. Advances in Carbohydrate Chemistry and Biochemistry.
2014;
71
:
9-136
.
View Article PubMed Google Scholar -
Kozakov
D.,
Hall
D.R.,
Xia
B.,
Porter
K.A.,
Padhorny
D.,
Yueh
C.,
The ClusPro web server for protein-protein docking. Nature Protocols.
2017;
12
(2)
:
255-78
.
View Article PubMed Google Scholar -
Weng
G.,
Wang
E.,
Wang
Z.,
Liu
H.,
Zhu
F.,
Li
D.,
HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Research.
2019;
47
:
322-30
.
View Article PubMed Google Scholar -
Laskowski
R.A.,
Thornton
J.M.,
PDBsum extras: SARS-CoV-2 and AlphaFold models. Protein Science.
2022;
31
(1)
:
283-9
.
View Article PubMed Google Scholar -
Krissinel
E.,
Henrick
K.,
Inference of macromolecular assemblies from crystalline state. Journal of Molecular Biology.
2007;
372
(3)
:
774-97
.
View Article PubMed Google Scholar -
Humphrey
W.,
Dalke
A.,
Schulten
K.,
VMD: visual molecular dynamics. Journal of Molecular Graphics.
1996;
14
(1)
:
33-8
.
View Article PubMed Google Scholar -
Phillips
J.C.,
Hardy
D.J.,
Maia
J.D.,
Stone
J.E.,
Ribeiro
J.V.,
Bernardi
R.C.,
Scalable molecular dynamics on CPU and GPU architectures with NAMD. The Journal of Chemical Physics.
2020;
153
(4)
:
044130
.
View Article PubMed Google Scholar -
Case
D.A.,
Aktulga
H.M.,
Belfon
K.,
Ben-Shalom
I.,
Brozell
S.R.,
Cerutti
D.S.,
Amber 2021University of California: San Francisco; 2021.
Google Scholar -
Duan
Y.,
Wu
C.,
Chowdhury
S.,
Lee
M.C.,
Xiong
G.,
Zhang
W.,
A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. Journal of Computational Chemistry.
2003;
24
(16)
:
1999-2012
.
View Article PubMed Google Scholar -
Grant
B.J.,
Skjaerven
L.,
Yao
X.Q.,
The Bio3D packages for structural bioinformatics. Protein Science.
2021;
30
(1)
:
20-30
.
View Article PubMed Google Scholar -
Aarthy
M.,
Singh
S.K.,
Envisaging the conformational space of proteins by coupling machine learning and molecular dynamics. InAdvances in Protein Molecular and Structural Biology Methods 2022 Jan 1 (pp. 467-475). Academic Press. .
View Article Google Scholar -
Hooft
R.W.,
Sander
C.,
Vriend
G.,
pubively judging the quality of a protein structure from a Ramachandran plot. Computer Applications in the Biosciences.
1997;
13
(4)
:
425-30
.
View Article PubMed Google Scholar -
Rahmani
F.,
Imani Fooladi
A.A.,
Ajoudanifar
H.,
Soleimani
N.A.,
In silico and experimental methods for designing a potent anticancer arazyme-herceptin fusion protein in HER2-positive breast cancer. Journal of Molecular Modeling.
2023;
29
(5)
:
160
.
View Article PubMed Google Scholar -
Istifli
E.S.,
Netz
P.A.,
Tepe
A. Sihoglu,
Husunet
M.T.,
Sarikurkcu
C.,
Tepe
B.,
In silico analysis of the interactions of certain flavonoids with the receptor-binding domain of 2019 novel coronavirus and cellular proteases and their pharmacokinetic properties. Journal of Biomolecular Structure & Dynamics.
2022;
40
(6)
:
2460-74
.
View Article PubMed Google Scholar -
Dar
A.M.,
Mir
S.,
Molecular docking: approaches, types, applications and basic challenges. Journal of Analytical {&}amp; Bioanalytical Techniques.
2017;
8
(2)
:
1-3
.
View Article Google Scholar -
Sun
Z.,
Li
Z.,
Zhao
L.,
Zhang
J.,
Research progress of anti-breast cancer peptides. International Journal of Innovative Research in Medical Science.
2019;
4
(08)
:
504-6
.
View Article Google Scholar -
Housman
G.,
Byler
S.,
Heerboth
S.,
Lapinska
K.,
Longacre
M.,
Snyder
N.,
Drug resistance in cancer: an overview. Cancers (Basel).
2014;
6
(3)
:
1769-92
.
View Article PubMed Google Scholar -
Pourhadi
M.,
Jamalzade
F.,
Jahanian-Najafabadi
A.,
Shafiee
F.,
Expression, purification, and cytotoxic evaluation of IL24-BR2 fusion protein. Research in Pharmaceutical Sciences.
2019;
14
(4)
:
320-8
.
View Article PubMed Google Scholar -
Jahanian-Najafabadi
A.,
Ghavimi
R.,
Akbari
V.,
10P In vitro and in vivo cytolethal and antitumor effects of a novel fusion protein targeting IL-24 toward breast cancer cells. Annals of Oncology : Official Journal of the European Society for Medical Oncology.
2020;
31
:
S2
.
View Article Google Scholar -
Soleimani
M.,
Mahnam
K.,
Mirmohammad-Sadeghi
H.,
Sadeghi-Aliabadi
H.,
Jahanian-Najafabadi
A.,
Theoretical design of a new chimeric protein for the treatment of breast cancer. Research in Pharmaceutical Sciences.
2016;
11
(3)
:
187-99
.
PubMed Google Scholar -
Muhammad
R. Hafiz,
Wardah
S.,
Naveed
K. Muhammad,
Numan
Y.,
Fareeha
B.,
Hamid
B.,
A Comprehensive In Silico Study of the NDB-IL-24 Fusion Protein for Tumor Targeting: A Promising Anti-Cancer Therapeutic Candidate. Journal of Biological Regulators and Homeostatic Agents.
2024;
38
(4)
:
3449-61
.
View Article Google Scholar -
Rehman
H.M.,
Rehman
H.M.,
Ahmed
N.,
Amirzada
M.I.,
Aslam
S.,
Bashir
H.,
HM
REHMAN,
In silico Design and Evaluation of Novel Cell Targeting Melittin-Interleukin-24 Fusion Protein: A Potential Drug Candidate Against Breast Cancer. Sains Malaysiana.
2023;
52
(11)
:
3223-37
.
View Article Google Scholar -
Xiao
B.,
Li
W.,
Yang
J.,
Guo
G.,
Mao
X.H.,
Zou
Q.M.,
RGD-IL-24, a novel tumor-targeted fusion cytokine: expression, purification and functional evaluation. Molecular Biotechnology.
2009;
41
(2)
:
138-44
.
View Article PubMed Google Scholar -
Gupte
T.M.,
Ritt
M.,
Sivaramakrishnan
S.,
ER/K-link-Leveraging a native protein linker to probe dynamic cellular interactions. Methods in Enzymology.
2021;
647
:
173-208
.
View Article PubMed Google Scholar -
Lee
C.D.,
Sun
H.C.,
Hu
S.M.,
Chiu
C.F.,
Homhuan
A.,
Liang
S.M.,
An improved SUMO fusion protein system for effective production of native proteins. Protein Science.
2008;
17
(7)
:
1241-8
.
View Article PubMed Google Scholar -
Agarwal
S.,
Chadha
D.,
Mehrotra
R.,
Molecular modeling and spectroscopic studies of semustine binding with DNA and its comparison with lomustine-DNA adduct formation. Journal of Biomolecular Structure & Dynamics.
2015;
33
(8)
:
1653-68
.
View Article PubMed Google Scholar
Comments
Article Details
Volume & Issue : Vol 12 No 2 (2025)
Page No.: 7138-7152
Published on: 2025-02-28
Citations
Copyrights & License
This work is licensed under a Creative Commons Attribution 4.0 International License.
Search Panel
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Google Scholar
Pubmed
Search for this article in:
Google Scholar
Researchgate
- HTML viewed - 1209 times
- PDF downloaded - 369 times
- XML downloaded - 69 times
