

Outsmarting the Immune System: The Complexities of Breast Cancer Immune Evasion from NK cells Surveillance
- Department of Biomedical Science, Advanced Medical and Dental Institute, Universiti Sains Malaysia, 13200 Bertam, Kepala Batas, Penang Malaysia
- Breast Cancer Translational Research Program@IPPT, BCTRP@IPPT, Advanced Medical and Dental Institute, Universiti Sains Malaysia, 13200 Bertam, Kepala Batas, Penang, Malaysia
Abstract
Breast cancer (BC) remains one of the leading causes of cancer-related deaths among women worldwide, largely attributable to its heterogeneity and capacity to evade immune surveillance. Natural killer (NK) cells are essential effectors of the innate immune system, mediating early recognition and destruction of malignant cells. However, BC employs multiple strategies to escape NK cell–mediated cytotoxicity. These include downregulation of activating ligands, upregulation of inhibitory receptors, altered cytokine and chemokine signaling, and the immunosuppressive effects exerted by the tumor microenvironment through stromal components and soluble mediators. This review article assesses both the therapeutic potential and limitations of targeting these evasion pathways. Finally, we discuss future directions in NK cell–based immunotherapy, including personalized approaches, advanced delivery systems, and combinatorial strategies aimed at promoting NK-cell survival, tissue infiltration, and cytotoxic function against tumors. A clear understanding of these evasion mechanisms is key to designing more effective treatments such as CAR-NK cells, checkpoint inhibitors, and cytokine-based therapies to reactivate the functionality of NK cells and improve clinical outcomes in breast cancer. This narrative review summarizes relevant studies identified through a structured search up to mid-2025, focusing on NK-cell biology, immune evasion, and therapeutic applications in breast cancer.
Background
Breast cancer (BC) is often characterized as a multifaceted disease due to its pronounced heterogeneity, which contributes to the emergence of diverse subtypes that exhibit distinct biological characteristics and responses to treatment 1. Heterogeneity manifests at various levels, including genetic, epigenetic, and phenotypic variations, which collectively drive the development of biologically diverse tumor subtypes and influence their therapeutic responses 2. The four subtypes of BC are classified according to genomic and receptor profiling: Luminal A (PR+, ER+, HER2−), Luminal B (PR+, ER+, HER2+), HER2-enriched (PR−, ER−, HER2+), and triple-negative breast cancer (TNBC; PR−, ER−, HER2−). Each subtype presents unique therapeutic challenges, particularly regarding recurrence and metastasis. Owing to the complexity and adaptability of breast tumors, conventional therapies often fail to achieve durable remission. In recent years, immunotherapeutic approaches have gained significant attention as potential treatment strategies, especially for challenging subtypes such as TNBC.
Natural killer (NK) cells have emerged as critical components of the innate immune system, recognized for their capacity to mediate cytotoxicity against tumor cells independent of antigen-specific priming 3. Despite this progress, our understanding of how breast cancer interacts with NK cells remains limited. While current research has largely emphasized adaptive immunity, focusing on T-lymphocyte-driven mechanisms, innate immune components—particularly NK cells—require further investigation.
Although substantial progress has been made in understanding adaptive immune evasion in BC, the mechanisms by which tumors specifically evade NK-cell surveillance remain underexplored. Most current immunotherapeutic strategies focus on T-cell modulation, overlooking the unique challenges and therapeutic opportunities associated with NK cells. A comprehensive understanding of the dynamic crosstalk between immune components and BC cells, particularly the mechanisms through which tumors evade NK-cell-mediated killing, is essential for developing more effective immunotherapeutic strategies. This review aims to address this gap by examining the key mechanisms utilized by BC to escape NK-cell-mediated immune surveillance and by exploring current and emerging strategies designed to enhance NK-cell cytotoxicity for BC management. A summary of immune surveillance mechanisms in BC is presented in Figure 1.
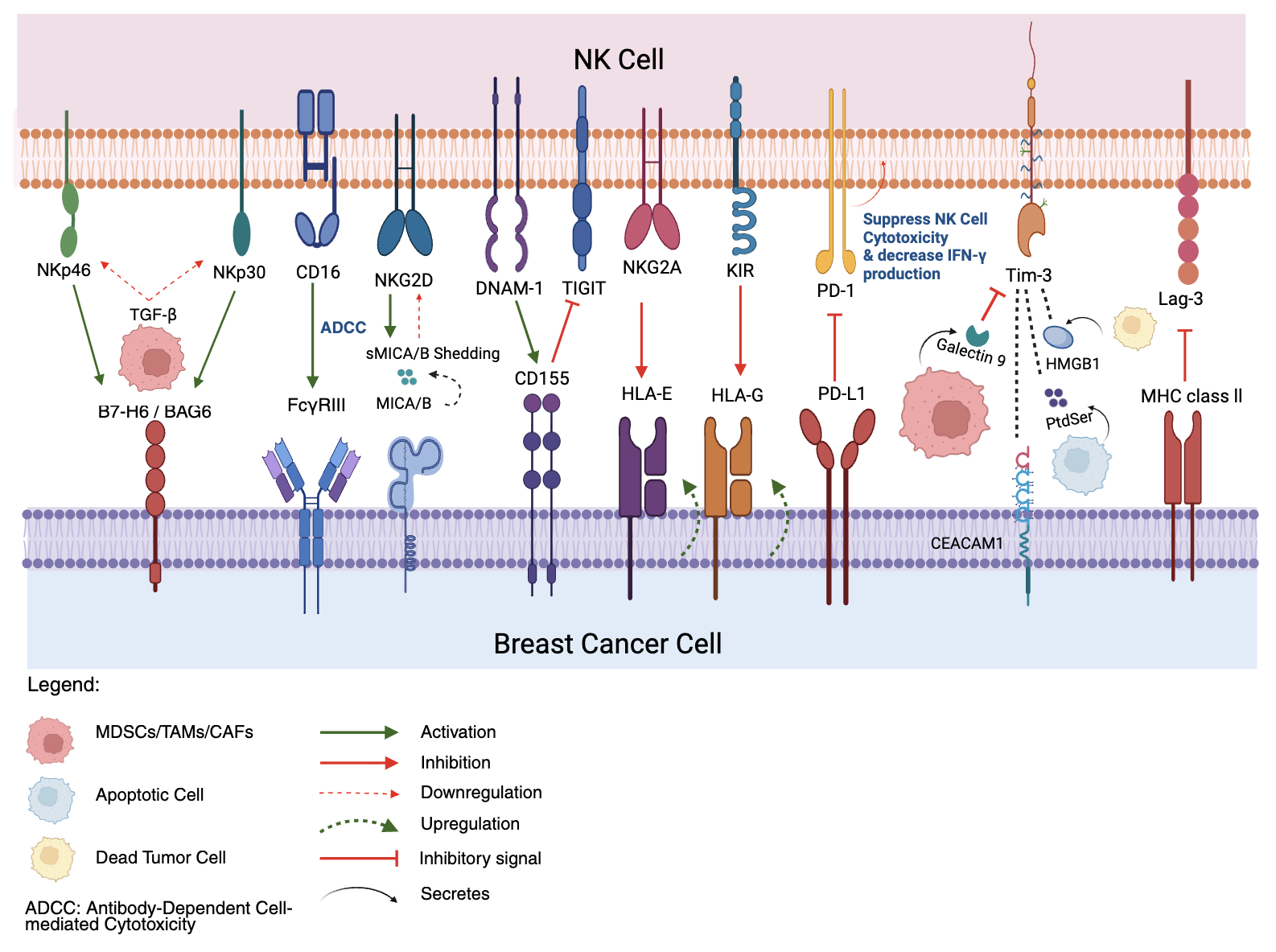
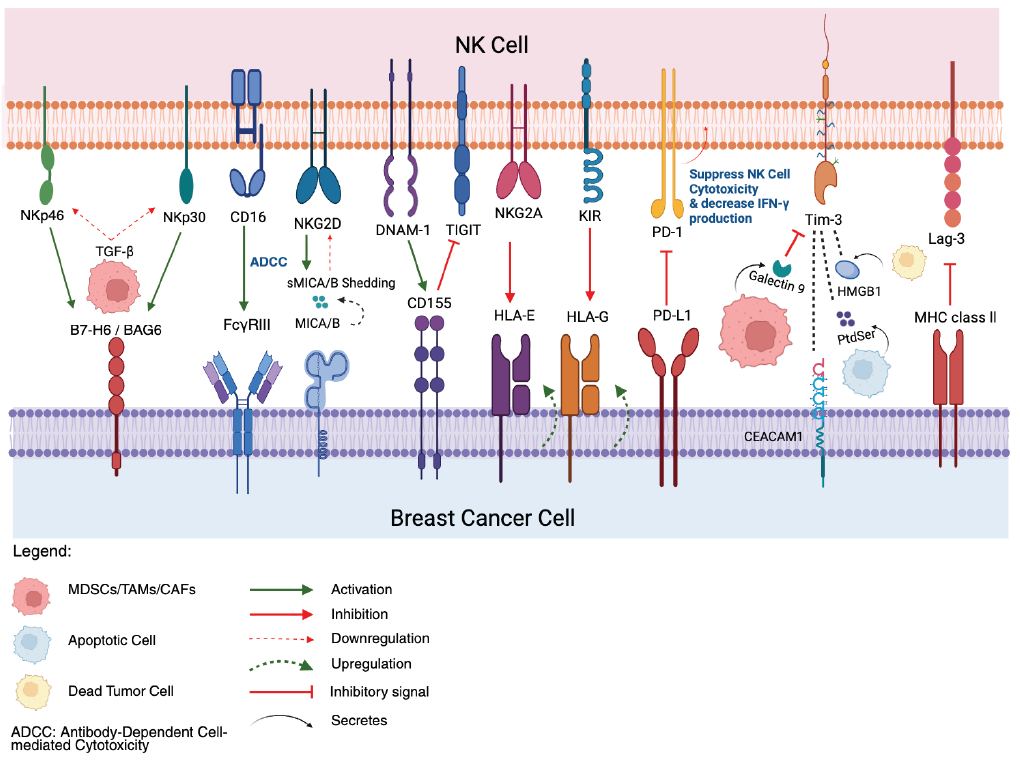
Breast Cancer Immune Evasion Mechanisms Targeting NK cells. Activating receptors on NK cells, such as NKG2D, DNAM-1, CD16, NKp46, and NKp30, typically recognize and bind to ligands (MICA/B, CD155, B7-H6/BAG6) on breast cancer cells, triggering NK cells activation and cytotoxicity. BC cells shed activating ligand, MICA/B (sMICA/B) to downregulate NK cells receptors and upregulate inhibitory ligands such as HLA-E and HLA-G to engage inhibitory NK cells receptors like NKG2A and KIRs, respectively. Breast cancer cells express immune checkpoint ligands (PD-L1, CD155, Galectin-9, HMGB1, MHC Class II, CEACAM1) that bind to their corresponding inhibitory receptors on NK cells (PD-1, TIGIT, Tim-3, Lag-3), leading to suppressed NK cells function, reduced cytotoxicity, and decreased IFN-γ production. The tumor microenvironment, populated by immunosuppressive cells like MDSCs, TAMs, and CAFs, further contributes to evasion by secreting inhibitory cytokines such as TGF-β which downregulates NKp30 and NKp46, creating an immunosuppressive microenvironment that impairs NK cells anti-tumor responses. Created in
Tumor Immunoediting and NK cells in BC
In breast cancer (BC), tumor immunoediting mediated by NK cells is a complex process that modulates immune surveillance and tumor progression. Research has highlighted the essential functions of NK cells in modulating immune activity in BC, particularly through mechanisms associated with tumor immunoediting. This concept underscores the cross-talk between immune and tumor cells, guiding their evolution through three sequential phases—elimination, equilibrium, and escape (Figure 2) 4. During the elimination phase, NK cells are activated by cellular stress signals and tumor-derived ligands. They exert cytotoxic effects by releasing perforin and granzymes and secrete pro-inflammatory cytokines, such as interferon-gamma (IFN-γ), thereby amplifying anti-tumor immunity.
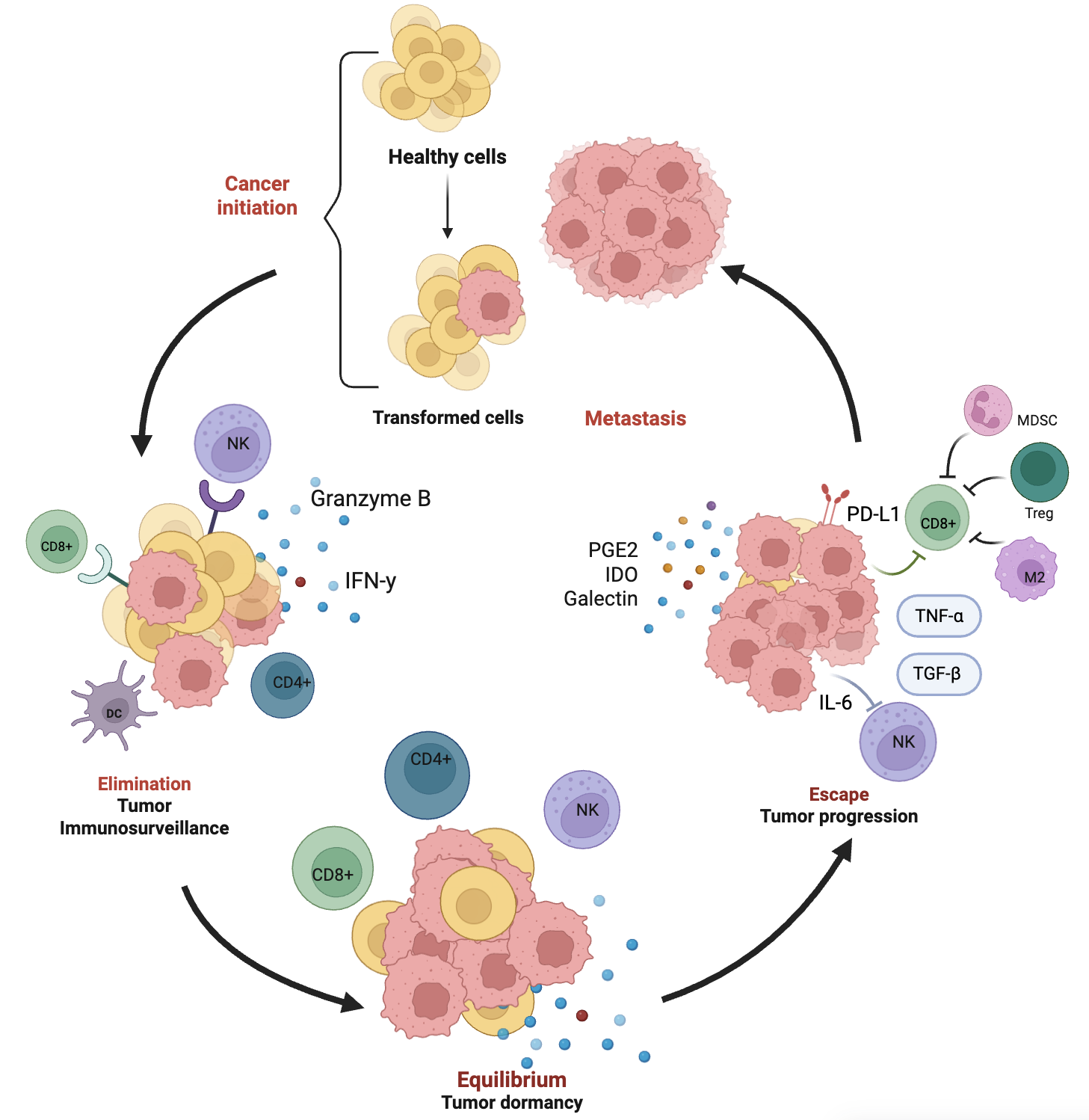
Tumor Immunoediting in Breast Cancer. The dynamic process of tumor immunoediting in breast cancer, comprising three key phases: elimination, equilibrium, and escape. During the elimination phase, transformed cells derived from healthy tissue are recognized and destroyed by the immune system, involving cytotoxic CD8+ T cells, CD4+ T cells, dendritic cells (DCs), and natural killer (NK) cells, which release effector molecules such as IFN-γ and granzyme B. In the equilibrium phase, residual tumor cells persist in a dormant state under continuous immune pressure, primarily maintained by CD4+ T cells and NK cells. In the escape phase, tumor cells acquire immune-evasive properties, aided by immunosuppressive cytokines (e.g., IL-6, TGF-β, TNF-α), and inhibitory molecules such as PD-L1. Immunosuppressive cells including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and M2-polarized macrophages (M2) contribute to the suppression of cytotoxic responses. Soluble mediators like PGE2, indoleamine 2,3-dioxygenase (IDO), and galectins further support immune evasion, leading to tumor progression and metastasis. Created in
In the equilibrium phase of immunoediting, tumor cells that survive the initial immune attack enter a state of dormancy, where their proliferation is balanced by immune-mediated destruction. Together with CD8 T cells and cytokines such as IFN-γ, NK cells help maintain this balance, exerting selective pressure that modulates the tumor’s immunogenic profile. The equilibrium phase is important because it allows for the selection of tumor variants with reduced immunogenicity, enabling them to persist in a latent state without overt progression 5.
During the escape phase, transformed cells that have gained immune-resistant features begin to proliferate uncontrollably, leading to clinically detectable disease. NK cell activity is often suppressed during this phase, either through the suppression of activating ligands, over-expression of inhibitory molecules, or the influence of immunoregulatory elements within the tumor microenvironment (TME), including regulatory T cells, myeloid-derived suppressor cells (MDSCs), and soluble factors like transforming growth factor-β (TGF-β) 6. These changes collectively impair NK-cell-mediated cytolytic activity and immunomodulatory cytokine secretion, allowing cancer cells to bypass immune recognition and drive metastatic progression 7.
In BC, NK cells are particularly important for identifying tumor cells that show reduced expression of major histocompatibility complex (MHC) class I molecules, a common mechanism used to escape T-cell-mediated immunity while concurrently exposing them to NK-cell surveillance 8. However, tumors that survive NK-cell surveillance enter the equilibrium phase. During this phase, NK–tumor cell interaction induces changes in the immune landscape which leads to the selection of immune-resistant variants with reduced immunogenicity 9. NK cells modulate the TME during this phase, helping to suppress excessive tumor growth 10. Eventually, some BC cells acquire additional immune-evasive traits, leading to the escape phase, during which tumor variants develop mechanisms to avoid NK-cell-mediated killing. These mechanisms include shedding or down-regulating activating ligands, up-regulating inhibitory ligands, or secreting immunosuppressive factors that impair NK-cell function. This allows tumors to establish an immunosuppressive TME and metastasize 11. Thus, understanding the interaction between NK cells and tumors is essential, as it highlights both their therapeutic potential and the mechanisms by which tumors evade the immune system.
Immune Evasion Mechanisms in BC
Loss/Alteration in Antigen Presentation
Tumor immune evasion in BC is a multifaceted process that profoundly affects antitumor immune responses, particularly those mediated by NK cells and cytotoxic T lymphocytes (CTLs). BC cells often escape immune surveillance by disrupting antigen presentation, most commonly through loss or functional alteration of major histocompatibility complex class I (MHC-I) molecules. This alteration diminishes the visibility of tumor cells to CTLs, enabling immune escape through several mechanisms, including genetic and epigenetic changes 12, reduced antigen expression 13, and transcriptional modulation 14. Although MHC-I-deficient cancer cells evade T-cell recognition, they are generally more susceptible to NK-cell-mediated cytotoxicity. Nevertheless, certain BC cells still avoid NK-cell detection owing to weak NK-cell activation and exhaustion 15.
Some BC cells exploit a phenomenon termed selective MHC-I expression, in which they retain non-classical MHC-I molecules such as HLA-E and HLA-G while down-regulating classical MHC-I 16. Engagement of these non-classical molecules with inhibitory receptors, including NKG2A and LILRB1, suppresses NK-cell activation and protects the tumor from NK-cell-mediated lysis 17,18. In addition, defects in the antigen-processing machinery (APM)—for example, loss of TAP1/2 or β2-microglobulin—impair peptide transport and loading onto MHC-I, thereby reducing tumor immunogenicity 19. Epigenetic silencing by DNA hypermethylation can likewise decrease the expression of MHC-I and related regulatory pathways; importantly, these changes are potentially reversible with DNA methyltransferase inhibitors (DNMTis) 20. Treatment with DNMTis such as guadecitabine, 5-azacytidine, or decitabine has been shown to restore MHC-I expression in BC 21. Moreover, combining DNMTis with pro-inflammatory cytokines (e.g., TNF-α or IFN-γ) or disabling NF-κB pathway inhibitors such as TNIP and N4BP1 further enhances MHC-I expression in BC cells 22,23,24.
Alterations in NK Cell Receptors and Ligands
NK cell activity is tightly controlled by a dynamic equilibrium between stimulatory and inhibitory signals, which are mediated through multiple receptors that detect both human leukocyte antigen (HLA) and non-HLA ligands on the surface of target cells, including tumor cells in breast cancer (BC). Disruption of this balance, particularly in the tumor microenvironment (TME), significantly reduces NK cell cytotoxic activity and eventually leads to immune evasion.
NK Cell Activating Receptors
Non-HLA-specific activating receptors include the natural cytotoxicity receptors (NCRs) NKp46, NKp30 and CD16 (FcγRIII), as well as Natural Killer Group 2D (NKG2D) and DNAX accessory molecule-1 (DNAM-1). In addition, certain killer cell immunoglobulin-like receptors (KIRs) and Natural Killer Group 2, member C (NKG2C) serve as HLA-specific activating receptors that promote NK cell effector functions upon engagement. In BC, these activating receptors are frequently down-regulated, specifically in tumour-infiltrating NK cells (Ti-NKs). Expression of NKG2D and NKp30 is reduced, which has been closely associated with impaired cytotoxic activity and poor immune surveillance, largely driven by immunosuppressive cues within the TME, highlighting the TME-mediated regulation of NK cell function and limiting antitumour responses in breast cancer 25,26. Furthermore, tumour-derived podocalyxin-like protein-1 (PCLP1), a CD34-family glycoprotein, has been shown to down-regulate the expression of activating receptors, leading to reduced NK cell activity and further hindering NK cell cytotoxicity in BC 27.
NK Cell Inhibitory Receptors
Counterbalancing these activating signals are several inhibitory receptors exploited by tumour cells to evade immune destruction. A key example is Natural Killer Group 2, member A (NKG2A), a C-type lectin-like receptor that forms a heterodimer with CD94 and interacts with its ligand HLA-E, a non-classical MHC class I molecule with limited polymorphism, thereby suppressing NK cell activity 28. Upon ligand engagement, immunoreceptor tyrosine-based inhibitory motifs (ITIMs) in NKG2A recruit phosphatases such as SHP-1, which suppress downstream activation pathways and inhibit NK cell cytotoxic function 29.
In the BC TME, both NKG2A and HLA-E are commonly up-regulated. Over-expression of HLA-E is driven by tumour cells and tumour-associated subsets, including CD141 conventional dendritic cells (DCs) and tumour-associated macrophages (TAMs) 30. This widespread expression enables tumour cells to engage NKG2A-expressing NK cells and deliver inhibitory signals that suppress NK cell activation, degranulation and cytokine production. Notably, Ti-NKs exhibit significantly higher levels of NKG2A compared with their circulating counterparts, suggesting that the TME promotes the up-regulation of inhibitory receptors to facilitate immune evasion 31.
NK Cell Activating Ligands
MHC class I-related chain A and B (MICA/B) and UL16-binding proteins (ULBPs) are important stress-induced ligands that engage the activating receptor NKG2D on NK cells. Their expression on tumour cells typically marks them for immune-mediated elimination. However, in BC, the expression of MICA/B is often down-regulated 32. The primary down-regulation mechanism of these ligands involves microRNAs (miRNAs), particularly those from the miR-17-92 cluster. Notably, miR-20a and miR-93, which regulate MICA/B, target the 3′-untranslated region (3′-UTR) of MICA/B mRNA, leading to transcript degradation and reduced protein expression 33. This post-transcriptional suppression diminishes NK cell activation, impairs NK cell-mediated cytotoxicity and enhances tumour growth, migration and invasion, thereby facilitating BC progression and metastasis 34.
NK Cell Inhibitory Ligands
In parallel, BC cells often up-regulate inhibitory ligands that bind to NK inhibitory receptors. HLA-E, a non-classical MHC class I molecule, acts as an inhibitory ligand that binds to the inhibitory receptor NKG2A, transmitting inhibitory signals through ITIMs that suppress NK cell activation and block NK cytotoxicity 35.
Moreover, alterations in the expression of another inhibitory ligand, HLA-G, in BC have been linked to reduced survival rates and an increased capacity of tumours to evade immune surveillance. HLA-G binds both KIR2DL4 and LILRB1 (at lower levels), initiating complex signalling that suppresses NK effector functions. KIR2DL4 contains both activating and inhibitory motifs, modulating NK cell activity depending on the context of HLA-G engagement 36. The molecular mechanisms underlying HLA-G over-expression in BC remain incompletely elucidated. To investigate this, Zhang et al. (2019) 38 conducted a study using MCF-7 BC cells in which they induced epigenetic modification, specifically DNA demethylation in the HLA-G promoter region, by targeting DNMT and TET enzymes with DMOG, a non-specific 2-oxoglutarate (2-OG)-dependent dioxygenase inhibitor. This process suppressed NK cell cytotoxicity and contributed to BC progression 36.
Expression of immune checkpoint molecules
Immune checkpoints control immune system activity, acting as gatekeepers that enable NK cells to modulate their function by preventing excessive activation. Checkpoints such as programmed death-1 (PD-1), cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), T-cell immunoglobulin and mucin domain-3 (TIM-3), lymphocyte activation gene-3 (LAG-3) and T-cell immunoglobulin and ITIM domain (TIGIT) maintain immunological homeostasis and preserve self-tolerance.
Programmed death-1 (PD-1)
The inhibitory receptor PD-1 is best known for modulating T-cell responses; however, it is also up-regulated on tumour-infiltrating NK cells (Ti-NKs) in breast cancer (BC) under the influence of immunosuppressive cytokines such as IL-10 and TGF-β within the tumour microenvironment (TME), and is associated with an exhausted NK phenotype 39. Interaction between PD-1 and its ligand PD-L1, which is frequently over-expressed by BC cells, tumour-associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSCs), suppresses NK-cell cytotoxicity and cytokine secretion, allowing tumour cells to evade immune destruction 40. PD-1 expression on human NK cells has similarly been linked to immune escape in melanoma, lung, pancreatic, bladder and ovarian cancers 39.
In early-stage BC, PD-1 is often co-expressed with other checkpoints, including CTLA-4, particularly on CD16CD56dim NK cells, suggesting a coordinated mechanism of immune evasion in aggressive subtypes such as HER2-positive or highly proliferative tumours 41. Pro-inflammatory mediators, notably IFN-γ, further augment PD-L1 expression in the TME, thereby amplifying PD-1-mediated suppression. Humanised mouse models (hu-CB-BRGS) of triple-negative BC (TNBC) demonstrate that PD-1 blockade can restore NK-cell function and promote tumour regression 42.
Cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4)
CTLA-4 is an immune checkpoint receptor mainly expressed on activated T cells and regulatory T cells (Tregs), where it inhibits activation by competing with CD28 for CD80/CD86 binding 43. Although CTLA-4 expression on NK cells is controversial, some studies indicate that IL-2 stimulation can induce its expression concomitant with IFN-γ production 43. CTLA-4 expression on CD8 tumour-infiltrating lymphocytes in BC has been correlated with favourable outcomes in luminal A and B subtypes 44. Intriguingly, higher CTLA-4 levels are detected more frequently in younger patients and in advanced-stage disease, implying a complex role in anti-tumour immunity 41.
T-cell immunoglobulin and mucin domain-3 (TIM-3)
TIM-3 is an inhibitory receptor expressed on multiple immune subsets, including NK cells 45. It is considered a marker of NK-cell maturation; the cytotoxic CD56dimCD16 subset displays higher TIM-3 levels than the immature CD56bright population, typically acquired together with killer-cell immunoglobulin-like receptors (KIRs) 43. Under chronic stimulation in the TME, persistent TIM-3 up-regulation on Ti-NKs is associated with diminished cytotoxicity, reduced IFN-γ production and, in some contexts, apoptosis, particularly in BC 46.
TIM-3 mediates inhibition through several ligands—most prominently Galectin-9 (Gal-9), but also phosphatidylserine (PtdSer), high-mobility-group box-1 (HMGB1) and carcinoembryonic-antigen-related cell-adhesion molecule-1 (CEACAM-1)—all of which trigger immunosuppressive cascades that disrupt NK-cell activation 47,48. Pro-inflammatory cytokines such as IL-2, IL-15 and IL-18, abundant in inflamed or tumourigenic tissues, further up-regulate TIM-3, particularly on immature NK cells 49,50. In early-stage BC, elevated TIM-3 on tumour-infiltrating lymphocytes correlates with increased PD-1 and PD-L1 expression, underscoring a cooperative mechanism of immune evasion 51.
T-cell immunoglobulin and ITIM domain (TIGIT)
TIGIT (also known as WUCAM, Vstm3 or VSIG9) is an inhibitory receptor present on NK cells, CD8 T cells and Tregs (Zhang et al., 2023). In BC, TIGIT engages its high-affinity ligand CD155 (poliovirus receptor, PVR), which is over-expressed on tumour and antigen-presenting cells in the TME 53. This interaction initiates inhibitory signalling that suppresses NK-cell activation, cytokine release and tumour lysis 46. TIGIT contains both an ITIM and an ITT-like motif; following CD155 binding, phosphorylation recruits SH2-domain phosphatases such as SHIP-1, thereby interfering with PI3K and MAPK pathways 52. The adaptor β-arrestin-2 further links phosphorylated TIGIT to SHIP-1, inhibits TRAF6 auto-ubiquitination and attenuates NF-κB signalling, resulting in reduced IFN-γ production 54. Consequently, NK-cell cytotoxicity, IFN-γ/TNF-α secretion and degranulation (CD107a) are diminished.
TIGIT also promotes suppression by out-competing the activating receptor DNAM-1 (CD226) for CD155 binding 28. In patients with advanced BC, tumour cells frequently over-express CD155, whereas NK cells exhibit increased TIGIT and decreased CD226, leading to an exhausted phenotype 55. In TNBC, high TIGIT expression on CD8 and CD4 TILs correlates with poor immune responses, highlighting TIGIT as a promising immunotherapy target 56.
Lymphocyte activation gene-3 (LAG-3)
LAG-3 (CD223) is a transmembrane protein of the immunoglobulin superfamily with potent inhibitory effects on T-cell immunity. Besides activated CD4/CD8 T cells, LAG-3 is expressed on NK cells, Tregs, B cells, invariant NKT cells, dendritic cells and TILs 53. By binding MHC class II molecules with higher affinity than CD4, LAG-3 negatively regulates immune responses 57. Although its function in NK cells is less defined, emerging evidence suggests that chronic activation, as in cancer, may up-regulate LAG-3 and dampen NK-cell effector functions.
Activation of signalling pathways
Breast cancer (BC) cells exploit oncogenic signalling pathways that impair natural killer (NK)-cell recognition and function. These pathways modulate the expression of ligands for the activating NK-cell receptor NKG2D (MICA/B) and suppress NK-cell effector functions, thereby enabling tumour immune evasion.
HER2/HER3-mediated activation of PI3K/AKT and RAS/MAPK pathways
Alimandi et al. (1997) demonstrated that stimulation of HER2/HER3 heterodimers by neuregulin-1β (NRG1β) leads to increased expression of MICA/B via downstream activation of both PI3K/AKT and RAS/MAPK pathways in the BC cell lines T-47D and MDA-MB-453. Although increased MICA/B expression should enhance NK-cell-mediated cytotoxicity via NKG2D engagement, tumour cells circumvent recognition by releasing soluble forms of MICA/B through proteolytic shedding. HER3 is also critical to this axis, as its knock-down significantly reduces NRG1β-induced MICA/B up-regulation 32.
PI3K/AKT-driven inactivation of GSK-3β
In addition to regulating MICA/B, the PI3K/AKT signalling axis contributes to NK-cell dysfunction associated with metastatic progression via the inactivation of glycogen synthase kinase-3β (GSK-3β). AKT phosphorylates GSK-3β at Ser9, leading to increased accumulation of reactive oxygen species (ROS) in BC cells. This subsequently reduces the phosphorylation of eIF2B at Ser535, a translation-initiation factor required for the expression of NKG2D and its ligands, including MULT-1 and RAE-1. Reduced expression of these molecules limits NK-cell recognition and cytotoxicity in BC. Conversely, studies have shown that active GSK-3β maintains higher Ser535-phosphorylated eIF2B levels and promotes the expression of NKG2D ligands, thereby enhancing NK-cell-driven tumour surveillance 59,60.
Impact of the Tumor Microenvironment (TME) on Immune Responses
Matrix Metalloproteinases (MMPs)
In breast cancer, NK cells display both anti-tumor activity and, in some cases, pro-tumor functions, highlighting their complex role in disease progression. CD56brightCD16+ NK cells have been shown to promote tumor progression by secreting matrix metalloproteinase-9 (MMP-9) and pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and angiogenin, thereby driving angiogenesis in breast cancer (BC) and immune suppression through the expansion of regulatory immune cells, impairment of T-cell infiltration, and induction of T-cell exhaustion 61,62. Metalloproteinases such as ADAM17 can be activated by cytokine stimulation (e.g., IL-2 and IL-15), cleaving CD16 from NK-cell surfaces. This shedding impairs antibody-dependent cellular cytotoxicity (ADCC), weakening NK-mediated tumor surveillance 63,64.
Cancer-Associated Fibroblasts (CAFs)
As key stromal cells within the BC TME, cancer-associated fibroblasts (CAFs) often impede NK-cell infiltration and activity 65, primarily through secretion of immunosuppressive mediators such as TGF-β, which down-regulates NK-cell receptor expression and effector functions 66. They also reduce production of cytotoxic molecules (perforin, granzyme B) and inhibit IFN-γ synthesis via the SMAD3–T-BET signalling axis. CAFs induce miR-183, which suppresses DAP12 67, and locally produce Dickkopf-related protein-1 (DKK1), a Wnt-pathway antagonist that attenuates NK activity 68,69. In addition, CAFs remodel the extracellular matrix via CXCL12/SDF-1 and MMPs, collectively creating barriers to NK-cell trafficking and cytotoxicity 70.
Myeloid-Derived Suppressor Cells (MDSCs)
MDSCs within the BC TME exert potent immunosuppression through the release of IL-6, IL-10 and TGF-β, which impairs the function of immune cells, including T cells 71,72 and NK cells 73,74, and through the generation of reactive oxygen/nitrogen species (ROS/RNS) 62. This biochemical suppression is reinforced by cellular crosstalk that down-regulates key NK-cell activation receptors, such as NKG2D and CD247, an essential signalling subunit of NKp30, NKp46 and FcγRIII/CD16. Co-culture experiments have shown that MDSC–NK-cell interactions result in diminished NK-cell degranulation capacity and reduced IFN-γ secretion 75. A central pathway involved in immature MDSC-mediated suppression is the STAT3 signalling cascade, which drives NF-κB-dependent production of indoleamine 2,3-dioxygenase (IDO), an immunosuppressive enzyme that suppresses NK-cell proliferation and cytotoxicity. Studies in murine models have shown that STAT3 inhibition restores NK-cell activation markers—including NKG2D, CD69, FasL, granzyme B, perforin and IFN-γ—and reduces tumor progression in BC 76,77.
Tumor-Associated Macrophages (TAMs)
TAMs constitute a major component of innate immune infiltrates in the solid-tumor TME and, in BC, can polarize into two principal phenotypes: pro-inflammatory, tumor-suppressive M1-like macrophages, and immunosuppressive, tumor-promoting M2-like macrophages 78. In TNBC, M2-like TAMs are linked to enhanced tumor-cell proliferation, metastatic potential and poor prognosis (59.0 % vs 40.4 %, p = 0.02) 62,78. They secrete IL-10, TGF-β, chemokines (CCL2, CCL17, CCL22), pro-angiogenic molecules such as VEGF-A, and matrix-remodelling enzymes, thereby facilitating tumor invasion, angiogenesis and metastasis 79. Similarly, epidermal growth factor receptor (EGFR) signalling supports macrophage recruitment and polarization, reinforcing immunosuppression in the TME 80,81.
Secretion of immunosuppressive molecules
Immunosuppressive molecules, also known as immunomodulatory molecules, are a heterogeneous group of soluble mediators derived from multiple cellular sources that modulate the immune microenvironment by balancing stimulatory and inhibitory signals. However, in breast cancer (BC) their production is often co-opted by tumour cells and other stromal components to evade immune surveillance and to create an environment conducive to tumour growth (Table 1).
Immunomodulatory Molecules Facilitating Immune Escape of Breast Cancer Cells from NK Cell Surveillance
| Cytokine | Tumor-Derived or TME-Derived Source | NK Cell Evasion Strategy | Mechanism of Action | Functional Consequence | Alteration in Breast Cancer | Key References |
|---|---|---|---|---|---|---|
| TGF-β | Tumor cells, Tregs, MDSCs | Receptor downregulation, functional reprogramming | Suppresses NKG2D, NKp30 expression; induces NK → ILC1 phenotype; inhibits mTOR/IFN-γ signaling | Loss of cytotoxicity; impaired IFN-γ secretion; metabolic exhaustion | ↑ Elevated | |
| IL-10 | Tregs, TAMs, B cells | Immune suppression via cytokine antagonism | Inhibits IFN-γ and TNF-α release; suppresses antigen-presenting cell function | Dampened immune stimulation and NK cell responsiveness | ↑ Elevated | |
| IL-6 | Tumor cells, CAFs, TAMs | STAT3-driven immune escape | Activates STAT3 in NK cells → inhibition of granule exocytosis; promotes Treg/MDSC expansion | Immune suppression; reduced degranulation | ↑ Elevated | |
| IL-8 (CXCL8) | Tumor cells, stromal cells | Recruitment of suppressor cells | Attracts MDSCs and neutrophils; modulates TME chemotaxis | Indirect NK inhibition via myeloid infiltration | ↑ Elevated | |
| IL-1β | Tumor cells, TAMs | Pro-tumoral inflammation | Drives chronic inflammatory TME; suppresses effective NK-TME interaction | Dysregulated inflammation, reduced NK efficacy | ↑ Elevated | |
| IL-2 | CD4+ T cells, dendritic cells | Deprivation of NK survival signals | Low IL-2 impairs NK proliferation and persistence in TME | Poor expansion and longevity of NK cells | ↓ Reduced | |
| IL-15 | DCs, monocytes | Disrupted homeostatic signaling | Activates P13K and MAPK pathway; mediates ADCC, release of perforin and granzymes | Weak activation, low IFN-γ, impaired cytotoxicity | ↔/Altered | |
| IL-33 | Tumor microenvironment | Unclear role | Induces IFN- production, increases PD1/PDL1 expression, suppresses NK cytotoxicity | NK inhibition via checkpoint induction | ↔/Altered | |
| IL-18 | Tumor microenvironment | Checkpoint-mediated suppression | Induces IFN production, increases PD1/PDL1 expression, suppresses NK cytotoxicity | Suppressed NK activity | ↔/Altered | |
| TNF-α | Tumor cells, NK cells | Pro-tumoral in chronic setting | Induces chronic NF-κB activation; upregulates suppressive surface ligands | Promotes tumor progression; inhibits effective NK response | ↑ Elevated | |
| IFN-γ | NK cells, T cells | Feedback suppression by tumor | Chronic IFN-γ exposure induces PD-L1 expression on tumor cells; enhances immunosuppressive feedback loop | Upregulation of immune checkpoints; desensitization of NK cytotoxicity | Context-dependent ↑ | |
| VEGF | Tumor cells, endothelial cells | Immune privilege through angiogenesis | Suppresses NK infiltration and function via vascular remodeling; promotes MDSCs | Spatial exclusion and functional suppression of NK cells | ↑ Elevated | |
| PGE2 | Tumor cells, CAFs, NK cells | Suppression via EP2/EP4 signaling | Downregulates IL-2-mediated LAK activity; suppresses IFN-γ and TNF-α via EP2/EP4; reduced NK activating receptors perforin/granzyme release | Impaired cytotoxicity, reduced cytokine release | ↑ Elevated |
Transforming Growth Factor-Beta, TGF-β
TGF-β acts at multiple levels: it down-regulates activating receptors such as NKG2D and NKp30 on NK cells 82; induces metabolic exhaustion characterised by mitochondrial dysfunction and reduced glycolysis, linked to impaired mTOR signalling, especially in metastatic settings 83,84,85; and thereby weakens tumour–NK-cell interactions, limiting NK-cell activation in advanced BC. TGF-β also inhibits IFN-γ production 86 and promotes the recruitment of immunosuppressive cells, providing an effective escape mechanism for BC 6. In metastatic BC, elevated systemic and local TGF-β correlates with impaired NK-cell bioenergetics, rendering them ineffective in tumour lysis 84,87.
Interleukin-10, IL-10
IL-10 is a cytokine with complex, sometimes contradictory, roles in cancer immunity. Although generally immunosuppressive and associated with poor clinical outcomes in BC 88, it also shows anti-tumour potential. IL-10 inhibits angiogenesis by down-regulating pro-angiogenic factors such as VEGF, IL-6, TNF-α and MMP-9 89. Experimental studies indicate that IL-10 can enhance NK-cell activation and cytolytic activity in a dose-dependent manner, thereby promoting target-cell destruction. Conversely, systemic IL-10 administration in wild-type mice suppresses antigen-specific immune responses, blunting the broader adaptive response 90. This includes suppression of key cytokines such as IL-2 and IFN-γ, both of which are essential for NK-cell functional activation 91.
Interleukin-15, IL-15
IL-15 generally promotes NK-cell-mediated cytotoxicity, antibody-dependent cellular cytotoxicity (ADCC) and cytokine production. It up-regulates activating receptors (e.g., NKG2D, NKp46) and cytolytic molecules (perforin, granzyme B, TRAIL) within the BC tumour microenvironment 92,93. Through the IL-2Rβ (CD122) and γc receptor subunits, IL-15 activates the JAK/STAT pathway. By limiting expression of the pro-apoptotic protein Bim and sustaining Mcl-1 expression—processes that involve Erk1/2 activity and the transcription factor Foxo3a—IL-15 preserves NK-cell viability in BC 94,95.
Interleukin-6, IL-6
IL-6 impairs NK-cell function via STAT3 activation and promotes oncogenesis through epithelial–mesenchymal transition (EMT) in ER-positive BC. IL-6 production is often triggered by IL-1β, which is over-expressed in luminal-type BC subtypes that otherwise do not secrete IL-6 96. STAT3 activation down-regulates natural cytotoxicity receptors (NCRs) on NK cells, reducing tumour-killing capacity 65,97. IL-6 not only fosters chronic inflammation but also expands MDSCs, which inhibit T-cell activity and secrete additional immunosuppressive mediators, forming a feedback loop that further suppresses NK-cell function and accelerates tumour progression 98.
Prostaglandin E2, PGE2
PGE2, a bioactive lipid synthesised by cyclo-oxygenase-2 (COX-2) in both immune and tumour cells, leads to NK-cell suppression within the BC microenvironment 99. It reduces IL-2-driven lymphokine-activated killer (LAK) cell activity via the EP2 receptor, one of four G-protein-coupled PGE2 receptors 100. Activated NK cells normally secrete IFN-γ to promote anti-tumour immunity; however, selective EP2 activation with agonists such as butaprost diminishes IFN-γ production by 66 %, whereas EP4 agonism produces an even greater (86 %) inhibition 101,102. Cancer-associated fibroblasts (CAFs) secrete higher levels of PGE2 than normal fibroblasts—particularly in response to NK-cell presence—creating an inhibitory loop 103,104.
Hypoxia and the Tumor Microenvironment
Hypoxia—a state of reduced oxygen availability—is a hallmark of the tumor microenvironment (TME) in breast cancer (BC) and other solid malignancies. It arises from the rapid proliferation of tumor cells together with a disorganized, inefficient vascular network that cannot satisfy their increased metabolic demand 105. This oxygen deprivation modulates multiple cellular pathways and stimulates angiogenesis, invasion, and immune evasion. Natural killer (NK) cells are particularly sensitive to hypoxic stress; under such conditions, they undergo profound metabolic reprogramming that compromises their viability, proliferation, and cytotoxicity 106.
Studies have demonstrated that hypoxic conditions down-regulate critical NK activating receptors, thereby directly impairing NK-mediated cytotoxicity 107,108. Another key immunosuppressive mechanism involves the adenosine pathway: hypoxia up-regulates CD73, an ecto-nucleotidase that converts extracellular ATP into adenosine which subsequently accumulates within the TME. Adenosine then binds to A2A receptors (A2ARs) expressed on NK cells, initiating a cascade of inhibitory signals that reduce the secretion of perforin, granzymes, and cytokines like IFN-γ 109. Moreover, hypoxia increases the expression of matrix metalloproteinase-7 (MMP-7) in tumour-associated macrophages (TAMs); MMP-7 cleaves Fas ligand (FasL) on NK cells, further impairing their cytotoxic capacity against BC 110.
NK-based Immunotherapeutic Approaches in Breast Cancer
Immune Checkpoint Blockade (ICB)
PD-1/PD-L1 Blockade
Emerging studies underscore the promise of immune checkpoint blockade (ICB) in rejuvenating exhausted T cells. PD-1/PD-L1 inhibition with anti-PD-1 and anti-PD-L1 monoclonal antibodies (mAbs) also enhances NK-cell–mediated antitumor responses in breast cancer (BC) 48,111. The IgG1 anti-PD-L1 mAb avelumab has been shown to enhance NK-cell functionality, particularly in triple-negative breast cancer (TNBC), by promoting the production of cytokines such as IFN-γ and TNF-α. It also mediates antibody-dependent cellular cytotoxicity (ADCC), and its efficacy is linked to PD-L1 expression levels, which correlate with increased therapeutic sensitivity 112,113,114,115.
TIM-3 and TIGIT Blockade
TIM-3 is an emerging immune checkpoint receptor whose function is comparable to that of PD-1 and CTLA-4 and represents a novel immunotherapeutic target in BC 116,117. Blocking TIM-3 with antibodies such as MBG453 or TSR-022 has shown preclinical promise by restoring NK-cell proliferation, persistence, and cytotoxicity 118,119. In addition, TIM-3 inhibition synergizes with other checkpoints, such as PD-1 blockade, suggesting that dual checkpoint inhibition may elicit a more robust NK-cell-based immune response against BC 120.
Another important checkpoint is TIGIT. Inhibitors of TIGIT are currently undergoing phase I/II clinical evaluation (e.g., NCT05867771). The bispecific antibody PM1022, which targets TIGIT and PD-L1, exerts multipronged antitumor effects by (i) depleting suppressive Tregs, (ii) mobilizing myeloid effector functions, and (iii) enhancing NK- and T-cell-mediated cytotoxicity 121.
NKG2A Blockade
NKG2A has emerged as another actionable checkpoint. Monalizumab, a first-in-class anti-NKG2A antibody, can reverse HLA-E–mediated inhibition and thereby enhance NK-cell function. Early clinical evaluation of monalizumab (NCT04307329) in HER2-positive BC suggests potential benefit, particularly in combination with trastuzumab, by augmenting NK-cell-mediated ADCC and overcoming resistance mechanisms. Consequently, the NKG2A–HLA-E axis represents a promising, druggable checkpoint pathway for NK-based immunotherapy in BC 122,123.
Antibody-Based Therapy
Recent advances highlight the potential of the afucosylated anti-HER2 antibody H2Mab-77-mG2a-f, which more efficiently recruits NK cells via FcγRIIa engagement, thereby boosting antibody-dependent cellular cytotoxicity (ADCC) against HER2+ BC. These findings underscore that mAb engineering, including bispecific formats, can directly enhance NK-cell recruitment and cytotoxicity in patients with BC 121.
More recently, DF1001, a HER2-targeted tri-specific NK-cell engager therapy (TriNKET), has been shown to recruit both NK and CD8+ T cells. In a phase I study (NCT04143711) involving patients with BC, DF1001 treatment led to increased NK- and T-cell infiltration, elevated pro-inflammatory cytokines, and durable clinical responses, with a clinical benefit rate of approximately 40 % and partial responses lasting up to 13 months. These data position TriNKETs as a promising next-generation antibody-based approach to overcome immune evasion and augment NK-driven anti-tumor immunity in BC 121,124.
The inhibitory checkpoint receptor CD85j (LILRB1/ILT2) is overexpressed on NK cells in TNBC, where it significantly impairs cetuximab-mediated ADCC. Functional blockade of CD85j with the HP-F1 mAb has been shown to restore NK cytotoxicity, with the degree of restoration correlating with CD85j expression levels, thereby selectively enhancing the lysis of TNBC cells without affecting HLA-I-negative targets 125.
Engineering of NK Cells / CAR-NK Cell Therapy
Adoptive cell therapy (ACT) is an advanced immunotherapeutic approach in which patient-derived T cells or natural killer (NK) cells are isolated, expanded ex vivo, and reinfused to potentiate anti-tumor immunity. A particularly promising ACT modality involves genetic modification of effector cells with chimeric antigen receptors (CARs) – synthetic receptors that redirect immune specificity toward tumour-associated antigens (TAAs) and thereby increase the precision and cytotoxic potency of the transferred cells (NCT02844335, clinicaltrials.gov; registered 1 January 2016).
In breast cancer (BC), HER2 and epidermal growth factor receptor (EGFR) are the best-characterised CAR-NK targets. HER2-directed CAR-NK cells suppress growth of HER2-positive BC xenografts and can overcome tumour-derived inhibitory signalling 126. These data have led to a phase I clinical trial (NCT04319757, clinicaltrials.gov; registered 18 March 2020) evaluating the safety and preliminary efficacy of HER2-CAR-NK cells in HER2-positive solid tumours, including BC.
Likewise, EGFR-targeted CAR-NK cells demonstrate potent pre-clinical activity against triple-negative BC models 127. Incorporation of co-stimulatory signalling domains into the CAR architecture further improves NK-cell expansion, in-vivo persistence and cytokine secretion, thereby augmenting therapeutic efficacy 128.
Cytokine Therapies
Cytokine-based therapy has the potential to reverse tumor-induced immunosuppression, thereby permitting breast cancer cells to be detected and eliminated by NK cells. Conventional interleukin-2 (IL-2) monotherapy is limited by significant toxicity and its propensity to expand regulatory T cells (Tregs), which suppress NK-cell function. Levin et al. (2012) engineered an IL-2 “superkine” with higher affinity for IL-2Rβ, thereby improving stability and efficacy while eliciting minimal toxicity compared with wild-type IL-2 129.
Interleukin-15 (IL-15) is particularly effective at promoting NK-cell proliferation and functional activation (Table 2). In clinical studies, systemic IL-15 administration induced a marked efflux of NK cells into the bloodstream, followed by hyperproliferation that produced a ten-fold expansion of the NK-cell compartment 130,131. Combinatorial approaches have further improved outcomes: IL-21 combined with IL-15 stimulates interferon-γ (IFN-γ) release, enhancing the tumor-killing capacity of NK cells 132,133,134.
Clinical Trials Investigating Immunotherapeutic Approaches Targeting NK Cells in Breast Cancer
| Clinical Trial ID | Study status/Phase | Condition | Drug | Enrollment |
|---|---|---|---|---|
| NCT04319757 | Completed / Phase 1 | HER2-positive Metastatic BC | ACE1702 (anti-HER2 oNK cells) | 12 |
| NCT02844335 | Completed / Phase 1 & 2 | Advanced BC | Cryosurgery and NK Immunotherapy | 60 |
| NCT02843204 | Completed / Phase 1 & 2 | HER2-positve BC | Anti-PD-1 Pembrolizumab and NK Immunotherapy | 110 |
| NCT02926196 | Active, not recruiting / Phase 3 | TNBC | Anti-PD-L1 antibody (Avelumab) | 474 |
| NCT03941262 | Completed / Phase 1 | Metastatic Cancer, Recurrent Cancer | SNK01 (autologous NK cells) + Avelumab or Pembrolizumab | 27 |
| NCT02614833 | Completed / Phase 2 | Metastatic Breast Carcinoma | Eftilagimod alpha + Paclitaxel | 242 |
| NCT05385705 | Recruiting / Phase 1 | Refractory Metastatic HER2+ BC | Trastuzumab and Pertuzumab | 20 |
| NCT05686720 | Not Yet Recruiting / Early Phase 1 | Advanced TNBC | SZ011 CAR-NK cell | 12 |
| NCT02839954 | Unknown / Phase 1 & 2 | TNBC | Anti-MUC1 CAR-pNK cells | 10 |
| NCT02843126 | Completed / Phase 1 & 2 | Recurrent BC | Trastuzumab + NK Immunotherapy | 30 |
| NCT06026657 | Recruiting / Phase 1 & 2 | Metastatic GD2+ HER2– BC | TGFβi NK Cells ± Naxitamab | 42 |
| NCT02536625 | Completed / Observational | Metastatic BC (HR+ / HER2–) | Everolimus (NK cell modulation) | 62 |
| NCT04143711 | Recruiting/ Phase 1& 2 | HER2+ solid tumor | HER2/CD16/NKG2D combined with pembrolizumab | 378 |
| NCT04307329 | Completed/ Phase 2 | HER2+ solid tumor | Combination of monalizumab and trastuzumab | 12 |
Because IL-15 monotherapy can induce dose-dependent toxicity 135, next-generation IL-15 derivatives and superagonists have been developed to sustain NK-cell activation with improved safety profiles (NCT02384954, NCT04136756, NCT04616196). The IL-15 superagonist complex ALT-803 (N-803) has demonstrated reduced toxicity in early-phase trials 136. Moreover, combining immune-checkpoint blockade with N-803 has shown promise in augmenting antitumor immune responses in ovarian cancer and may represent a novel strategy for breast cancer (BC), which currently lacks an FDA-approved immune-checkpoint inhibitor 137,138.
Other Therapies
Epigenetic modulators such as histone deacetylase inhibitors (HDACis) and DNA methyltransferase inhibitors (DNMTis) can reverse epigenetically mediated silencing of the antigen-processing machinery (APM). A study exploring the combination of these epigenetic agents with immune-checkpoint blockade demonstrated that restoration of major histocompatibility complex (MHC) class I expression sensitises breast cancer (BC) cells to immunotherapy 34,139.
Beyond epigenetic reprogramming, several signalling pathways that contribute to natural killer (NK)-cell dysfunction have been proposed as therapeutic targets; for example, inhibition of the PI3K/Akt/GSK-3β/ROS/eIF2B axis may enhance NK-cell function in BC, although its clinical tractability is still under early investigation. Combining NK-cell-based immunotherapy with GSK-3β or NOX4 inhibitors, which regulate reactive oxygen species (ROS) production, may further improve treatment outcomes 59. Blockade of a disintegrin and metalloproteinases (ADAMs) with neutralising antibodies significantly reduces tumour-cell proliferation, underscoring their role in tumour growth and epidermal growth factor receptor (EGFR) signalling—pathways already exploited in BC therapy 140.
Finally, a combinatorial strategy employing EGFR-chimeric antigen receptor (CAR) NK cells—engineered to recognise EGFR-expressing BC cells—together with a self-replicating oncolytic herpes simplex virus type 1 (oHSV-1) has been designed to enhance therapeutic efficacy. This approach enables oHSV-1 to lyse residual cancer cells that are EGFR-negative or EGFR-low, which would otherwise evade NK-cell detection. Moreover, oHSV-1 up-regulates activating ligands and promotes NK-cell infiltration and functional activation within the tumour microenvironment (TME) 128,141.
Challenges and Future Directions
Despite the promising advances in immunotherapy, several barriers still hinder the clinical translation of NK-cell-based treatments in breast cancer (BC). A major obstacle is the tumor’s intrinsic and adaptive resistance to NK-cell-mediated cytotoxicity. BC cells can down-regulate activating ligands, secrete immunosuppressive cytokines, and up-regulate inhibitory checkpoints such as HLA-E and PD-L1. Prolonged therapeutic pressure may further induce receptor shedding or NK-cell exhaustion, thereby diminishing long-term efficacy. Tumor heterogeneity introduces an additional layer of complexity; distinct BC subtypes, such as triple-negative breast cancer (TNBC) and HER2-positive disease, display variable expression of NK-activating or inhibitory ligands. This heterogeneity not only modulates immune surveillance but also affects responsiveness to immunotherapy.
Another critical challenge is the limited lifespan, poor persistence, and insufficient tumor infiltration of adoptively transferred NK cells within the hostile tumour microenvironment (TME). This limitation is exacerbated by the accumulation of immunosuppressive cells, including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumour-associated macrophages (TAMs), which inhibit NK-cell function through direct cell–cell interactions and soluble mediators.
Multiple innovative strategies are under investigation to overcome these escape mechanisms and enhance targeting specificity, including next-generation chimeric antigen receptor (CAR)-NK cells, bispecific or trispecific killer engagers (BiKEs/TriKEs), and oncolytic viruses. Furthermore, engineering cytokine-fusion proteins or extracellular-vesicle-encapsulated cytokines may sustain NK-cell activation within solid tumours. A deeper understanding of the intricate interactions between NK cells and other immune populations will support the rational design of combination regimens. Finally, the integration of multi-omics datasets with artificial intelligence (AI) has the potential to accelerate biomarker discovery and facilitate patient stratification for personalised immunotherapy.
Conclusion
In summary, tackling the multifaceted immune evasion strategies employed by BC requires a multidisciplinary and patient-specific approach, posing a major challenge to immunotherapy. Continued research into the molecular drivers of NK-cell dysfunction, along with innovation in therapeutic design, will be essential for maximizing the therapeutic efficacy of NK-cell–based therapies. Advances in cytokine engineering, checkpoint inhibition, and CAR-NK-cell therapies offer promising solutions, but success will depend on personalized, multimodal approaches that reprogram both the tumor and its microenvironment.
Abbreviations
A2ARs: A2A receptors; ADAM17: A Disintegrin and Metalloproteinase 17; ADAMs: A Disintegrin and Metalloproteinases; ADCC: Antibody-dependent cellular cytotoxicity; AKT: Protein kinase B; APM: Antigen-processing machinery; BC: Breast cancer; BiKEs: Bispecific killer engagers; CAFs: Cancer-associated fibroblasts; CAR: Chimeric antigen receptor; CAR-NK: Chimeric antigen receptor–engineered natural killer cell; CD85j: Cluster of Differentiation 85j; CD94: Cluster of Differentiation 94; CEACAM-1: Carcinoembryonic-antigen-related cell-adhesion molecule 1; CTLs: Cytotoxic T lymphocytes; CTLA-4: Cytotoxic T-lymphocyte–associated antigen-4; DCs: Dendritic cells; DKK1: Dickkopf-related protein 1; DNAM-1: DNAX accessory molecule-1; DNMTs: DNA methyltransferases; DNMTis: DNA methyltransferase inhibitors; DMOG: Dimethyloxalylglycine; EGFR: Epidermal growth factor receptor; EMT: Epithelial–mesenchymal transition; EP2: Prostaglandin E₂ receptor 2; EP4: Prostaglandin E₂ receptor 4; ER: Estrogen receptor; FasL: Fas ligand; FOXO3a: Forkhead box O3a; GSK-3β: Glycogen synthase kinase-3 beta; HMGB1: High-mobility-group box-1; HLA: Human leukocyte antigen; HLA-E: Human leukocyte antigen-E; HLA-G: Human leukocyte antigen-G; hu-CB-BRGS: Human cord-blood-reconstituted BRGS mouse; ICB: Immune checkpoint blockade; IDO: Indoleamine 2,3-dioxygenase; IFN-γ: Interferon-gamma; IL-1β: Interleukin-1 beta; IL-2: Interleukin-2; IL-6: Interleukin-6; IL-10: Interleukin-10; IL-15: Interleukin-15; IL-18: Interleukin-18; IL-21: Interleukin-21; ILT2: Immunoglobulin-like transcript 2; ITIMs: Immunoreceptor tyrosine-based inhibitory motifs; KIRs: Killer cell immunoglobulin-like receptors; KIR2DL4: Killer cell immunoglobulin-like receptor 2DL4; LAK: Lymphokine-activated killer; LAG-3: Lymphocyte activation gene-3; LILRB1: Leukocyte immunoglobulin-like receptor subfamily B member 1; MAPK: Mitogen-activated protein kinase; MDSCs: Myeloid-derived suppressor cells; MHC: Major histocompatibility complex; MHC-I: Major histocompatibility complex class I; MICA/B: MHC class I-related chain A and B; miRNAs: MicroRNAs; mAbs: Monoclonal antibodies; mRNA: Messenger ribonucleic acid; mTOR: Mechanistic target of rapamycin; N4BP1: NEDD4-binding protein 1; NCRs: Natural cytotoxicity receptors; NF-κB: Nuclear factor κB; NKG2A: Natural Killer Group 2 member A; NKG2C: Natural Killer Group 2 member C; NKG2D: Natural Killer Group 2D; NK: Natural killer cells; NKp30: Natural cytotoxicity receptor p30; NKp46: Natural cytotoxicity receptor p46; NOX4: NADPH oxidase 4; oHSV-1: Oncolytic herpes simplex virus type 1; PD-1: Programmed death-1; PD-L1: Programmed death ligand-1; PGE₂: Prostaglandin E₂; PI3K: Phosphoinositide 3-kinase; PR: Progesterone receptor positive; PtdSer: Phosphatidylserine; PVR: Poliovirus receptor; RAE-1: Retinoic acid early inducible-1; RNS: Reactive nitrogen species; ROS: Reactive oxygen species; SHP-1: Src homology region 2 domain-containing phosphatase-1; SHIP-1: SH2 domain-containing inositol 5′-phosphatase 1; SMAD3: Mothers against decapentaplegic homolog 3; STAT3: Signal transducer and activator of transcription 3; sMICA/B: Soluble MHC class I-related chain A and B; TAA: Tumor-associated antigen; TAMs: Tumor-associated macrophages; T-BET: T-box transcription factor TBX21; TET: Ten-eleven translocation dioxygenase; TGF-β: Transforming growth factor-beta; TIGIT: T-cell immunoglobulin and ITIM domain; TIM-3: T-cell immunoglobulin and mucin domain-3; Ti-NKs: Tumor-infiltrating natural killer cells; TNF-α: Tumor necrosis factor-alpha; TNBC: Triple-negative breast cancer; TRAF6: TNF receptor–associated factor 6; TRAIL: TNF-related apoptosis-inducing ligand; TriNKETs: Tri-specific NK-cell Engager Therapy; Tregs: Regulatory T cells; TME: Tumor microenvironment; ULBPs: UL16-binding proteins; VEGF: Vascular endothelial growth factor; VEGF-A: Vascular endothelial growth factor-A.
Acknowledgements
None.
Author’s Contributions
LS contributed to the conception. LS and RS wrote the manuscript and generated the figures. HH and SRAR review the manuscript. NH participated in drafting the review and revising it critically for important intellectual content. All authors reviewed the final manuscript submitted. All authors carefully evaluated and endorsed the final manuscript for submission to the journal.
Funding
This project is funded by the National Cancer Council (MAKNA Cancer Research Award 2023) and BCTRP@IPPT (1001/CIPPT/8070033).
Declaration of generative AI and Ai-assisted technologies in the writing process
The authors utilized OpenAI’s ChatGPT to support the refinement of language, organization of content, and formatting throughout the development of this manuscript. All outputs generated were carefully reviewed and modified by the authors to ensure accuracy and integrity. The authors accept full responsibility for the final content presented.
Competing Interests
The authors declare that they have no competing interest.
Ethical considerations
None applicable.