

The SDHD H102R Variant is a Genetic Risk Factor for Head and Neck Paraganglioma in the Russian Population
- Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, 119991 Moscow, Russia
- Vishnevsky Institute of Surgery, Ministry of Health of the Russian Federation, 117997 Moscow, Russia
Abstract
Introduction: Head and neck paraganglioma (HNPGL) is an extremely rare tumor with a strong genetic predisposition. Approximately 40% of HNPGLs are associated with mutations in the SDHx genes, which can harbor pathogenic genetic alterations specific to certain populations. In our previous research, we identified a frequent genetic variant—NM_003002: c.305A>G, p.H102R (chr11:111959726, rs104894302)—in the SDHD gene among Russian patients diagnosed with HNPGL. The present study analyzed expanded case and control cohorts to accurately estimate the association between this variant and the risk of developing the disease.
Methods: The study comprised two expanded cohorts: 187 patients with HNPGL and 1,055 healthy individuals of Russian descent. Genetic testing for the SDHD H102R variant was performed using targeted sequencing on an Illumina platform.
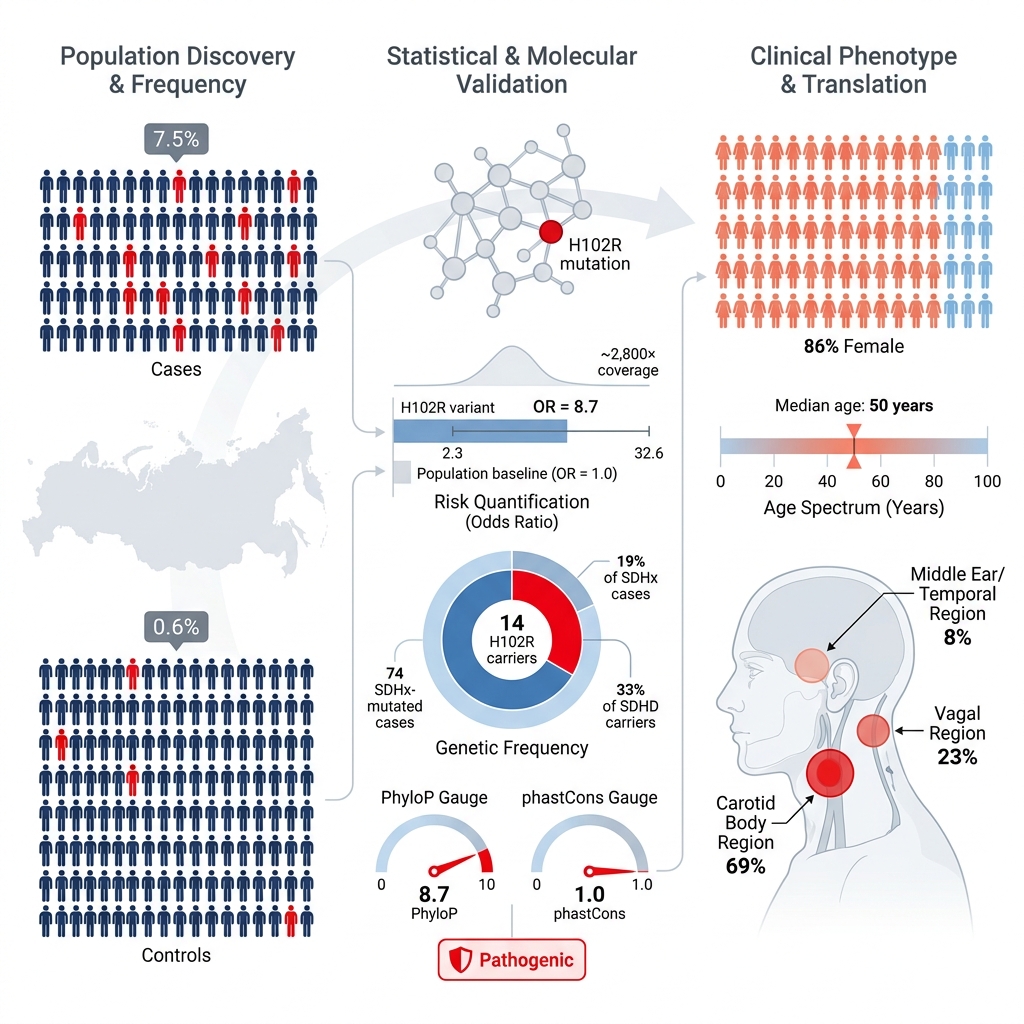
Results: The SDHD H102R variant was detected in 7.5% of the overall patient cohort and in 19% of all SDHx-mutated cases. Furthermore, the variant was present in one-third of all SDHD mutation carriers. In the control cohort, the frequency of the SDHD H102R variant was 0.6%. The adjusted odds ratio, accounting for sex and age covariates, was 8.7 (95% CI: 2.3–32.6, p = 0.001).
Conclusion: This case-control study demonstrates a strong association between the SDHD H102R variant and the risk of developing HNPGL in the Russian population.
Introduction
Head and neck paraganglioma (HNPGL) is a rare neuroendocrine neoplasm arising from the neuroendocrine cells of parasympathetic paraganglia 1. HNPGL exhibits a high degree of heritability (up to 40%) and is predominantly caused by mutations in the succinate dehydrogenase (SDHx) genes. Somatic SDHx mutations are rarely observed in apparently sporadic HNPGLs and have been predominantly identified within the SDHD gene 2,3. Clinically, SDHx mutations are of particular significance, as they predispose individuals to a variety of clinical presentations, including hereditary paraganglioma (PGL) syndromes (all SDHx mutations), multifocality (SDHD), and malignancy (SDHB) 4. Consequently, genetic testing for SDHx mutations is imperative for the clinical management of patients with PGLs and for the surveillance of their at-risk family members.
The spectrum of genetic variants within the SDHx genes varies significantly across diverse populations, with several mutations demonstrating strong founder effects or population-specific prevalence. Examples include an SDHB deletion in Brazil, the Netherlands, and Spain 5,6; SDHC p.Arg133Ter in the French Canadian population 7; SDHD p.Asp92Tyr, p.Leu95Pro, and p.Leu139Pro in the Netherlands 8,9; SDHD p.Cys11Ter in Central Europe; SDHD p.Gln109Ter and p.Tyr114Cys in Italy 10; and SDHD p.Pro81Leu in the Americas 11. Similarly, a highly prevalent SDHD H102R variant has been identified in Russian patients with HNPGLs 12.
In this study, we performed a comprehensive analysis of the SDHD H102R variant frequency in Russian patients with HNPGLs and an ethnically matched control population to robustly characterize its clinical significance. Compared to our previous study 12, the current investigation utilizes expanded cohorts for both HNPGL patients (n = 187) and healthy controls (n = 1,055) subjected to genetic testing. This substantial increase in cohort size enhances the statistical power required for the accurate clinical classification of this mutation, thereby facilitating a more precise determination of its population-specific frequency and pathogenicity.
Methods
Study subjects
A total of 187 patients with HNPGLs (contributing 204 tumor samples and 11 normal tissue samples) were included in the study 12. Notably, patients with middle ear paragangliomas—a subset of HNPGLs not previously investigated in this context—were also included. Formalin-fixed, paraffin-embedded (FFPE) tumor samples were collected following histopathological examination at the Pathology Department of the Vishnevsky Institute of Surgery. The clinicopathological characteristics of the patient cohort are summarized in Table 1.
The ethnically matched control group comprised 1,055 unrelated Russian individuals with no history of cancer. The control cohort exhibited a 1:1 female-to-male ratio and a median age of 71 years (mean ± SD = 72.3 ± 8.5 years). Normal tissue samples from healthy Russian adults were provided by the Laboratory of Biological Microarrays at the Engelhardt Institute of Molecular Biology. Written informed consent was obtained from all participants for the molecular genetic analysis and the subsequent publication of the findings. The study protocol was approved by the Ethics Committee of the Vishnevsky Institute of Surgery and was conducted in accordance with the Declaration of Helsinki.
The clinicopathologic characteristics of 187 Russian patients with head and neck paragangliomas (representing 204 tumors). The following table provides a description of the case cohort (n = 187) that were subjected to genetic testing on the
| Characteristics | Data |
|---|---|
| Total, n | |
| Patients | 187 |
| Tumors | 204 |
| Age, years | |
| Range (Min–Max) | 16 – 84 |
| Mean ± SD / Median | 49.7 ± 13.7 / 50 |
| Sex, n patients (%) | |
| Female | 143 (76.5%) |
| Male | 44 (23.5%) |
| Localization, n tumors (%) | |
| Carotid paraganglioma | 128 (63%) |
| Vagal paraganglioma | 38 (19%) |
| Middle ear paraganlioma | 27 (13%) |
| Unknown | 11 (5%) |
| Tumor clinical course, n patients (%) | |
| Recurrent | 9 (5%) |
| Metastatic | 7 (4%) |
| Multifocal | 14 (7.5%) |
| No | 113 (60%) |
| Any | 74 (40%) |
| 3 (2%) | |
| 23 (12%) | |
| 8 (4%) | |
| 42 (22.5%) | |
DNA Isolation
Genomic DNA was extracted from FFPE tumor tissues (case cohort) and peripheral blood (control cohort) using the High Pure FFPET DNA Isolation Kit (Roche, Basel, Switzerland) and the MagPure Blood DNA Kit (Magen Biotech, Guangzhou, China), respectively, according to the manufacturers' instructions. Additionally, DNA was isolated from matched normal tissues (blood or lymph nodes) of patients with identified SDHD H102R mutations using the aforementioned kits. DNA quantification was performed using a Qubit 4 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
High-Throughput Targeted Sequencing
Genetic testing for the SDHD H102R variant was conducted via high-throughput targeted sequencing. Amplicon libraries were prepared using a two-stage polymerase chain reaction (PCR) protocol. The first stage utilized primers specific to the target locus within the SDHD gene, while the second stage employed dual-index barcoding primers. The primer sequences for the first-stage PCR were as follows: forward, 5'-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGTTTAGGGCATTTCAATCAACTTCTC-3'; and reverse, 5'-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGACTAGCGAGAGGGTTGTC-3'. This primer set demonstrates high specificity for the SDHD H102R target locus (NC_000011.10, Homo sapiens chromosome 11, GRCh38.p14 Primary Assembly), yielding a 287 bp amplicon with no off-target amplification. In the second PCR stage, Nextera XT i5 and i7 indexing primers (Illumina, San Diego, CA, USA) were used for multiplexing, resulting in a final library length of approximately 370 bp.
All PCR reactions were performed using the Tersus Plus PCR kit (Evrogen, Moscow, Russia) on a T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA). The first-stage PCR thermocycling conditions were: 95 °C for 3 min; 30 cycles of 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 30 s; followed by a final extension at 72 °C for 5 min and a 4 °C hold. The second-stage amplification conditions were: 95 °C for 3 min; 8 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s; followed by a final extension at 72 °C for 5 min and a 4 °C hold. Following each PCR step, amplicons underwent size verification via 2% agarose gel electrophoresis and purification using MagPure beads (Magen Biotech) in accordance with the manufacturer's protocol. The final libraries were equimolarly pooled and sequenced on an Illumina MiSeq System using a 2 × 151 bp paired-end configuration. A minimum of 2,000 reads was obtained per sample.
Bioinformatics analysis commenced with standard read quality control (Q ≥ 30) using FastQC v0.11.5 13 (, accessed on 06 October 2025). Trimmomatic v0.39 was utilized to remove adapter sequences and short reads (<40 bp) 14. Reads were aligned to the GRCh37.p75 human reference genome using the BWA-MEM aligner v2.2.1 15. Variant calling was performed using two independent algorithms: freeBayes v1.3.2 (diploid genotype parameters: --min-alternate-fraction 0.2 --min-alternate-count 3) 16 and GATK HaplotypeCaller v4.2.4.0 (default parameters for germline short variant calling) 17. Stringent quality filters were applied during variant calling with both tools; only reads with a minimum mapping quality of ≥ 30 and bases with a minimum base quality of ≥ 30 were retained. Following filtration, the mean amplicon coverage was approximately 2800×.
For the resulting variants, the variant allele frequency (VAF) was calculated as the ratio of alternative allele reads to the total reads (alternative plus reference). High-confidence germline variants were selected based on VAF thresholds: variants with a VAF between 0.35 and 0.65 were classified as heterozygous, whereas those with a VAF ≥ 0.9 were classified as homozygous alternative. Variant interpretation was facilitated by multiple resources, including allele frequency databases (1000 Genomes Project, ExAC, gnomAD, Kaviar, and ESP-6500), variant databases (dbSNP, ClinVar, and COSMIC), evolutionary conservation algorithms (PhastCons and PhyloP), and the InterPro database for protein domain localization. Additionally, several in silico pathogenicity prediction algorithms were utilized, including SIFT, PolyPhen-2, MutationTaster, LRT, InterVar, PROVEAN, MetaSVM, MetaLR, CADD, DANN, and FATHMM, among others.
Sanger Sequencing
To determine the germline or somatic status of the identified SDHD H102R variants, Sanger sequencing was performed on genomic DNA extracted from matched normal tissues. Genomic DNA was amplified using the aforementioned SDHD H102R locus-specific primers, PCR master mix, and thermocycling conditions (as described in the first-stage PCR protocol for targeted library preparation). PCR products were subsequently purified using MagPure beads (Magen Biotech, Guangzhou, China) in accordance with the manufacturer's instructions. Sequencing was executed on an ABI 3500xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) utilizing the BigDye Terminator v3.1 Cycle Sequencing Kit. The resulting sequence data were analyzed using DNASTAR software (DNASTAR, Madison, WI, USA).
Statistical analysis
The association between the SDHD H102R variant and HNPGLs was assessed. The 95% confidence interval (CI) for the mutation frequency was calculated using the standard formula for a binomial proportion (percentage of a characteristic):
where p is the sample proportion, n is the total sample size, and z =1.96 for a 95% confidence level.
The strength of the association was quantified by calculating the unadjusted (crude) odds ratio (OR) from a 2×2 contingency table. Statistical significance was assessed using Pearson's chi-square test. The 95% CI for the OR was derived using the Woolf (logit) method:
where a, b, c, and d represent the cell frequencies (variant carriers with disease, carriers without disease, non-carriers with disease, and non-carriers without disease, respectively).
To control for potential confounding factors, an adjusted odds ratio (aOR) was calculated using multiple logistic regression with the Statsmodels package in Python, incorporating sex and age as covariates.
Results
The target variant NM_003002: c.305A>G, p.H102R (chr11:111959726, rs104894302) in exon 3 of the SDHD gene was detected as a heterozygous mutation in 14 of 187 (7.5%; 95% CI: 3.7–11.3) patients with HNPGLs. The frequency of the SDHD H102R variant was 19% (14/74; 95% CI: 9.9–27.8) among SDHx mutation carriers and 33% (14/42; 95% CI: 19.0–45.7) among SDHD mutation carriers specifically. Germline versus somatic mutational status was evaluated in 11 patients for whom matched normal tissues were available. In the majority of these cases (10 of 11), the SDHD H102R variant was of germline origin. The acquisition of additional normal tissue samples was precluded by the retrospective nature of the study and the reliance on archived tumor specimens.
Patients harboring the SDHD H102R variant were predominantly female (86%, 12/14), with a median age of 50 years. The majority presented with carotid paragangliomas (69%), followed by vagal (23%) and middle ear (8%) paragangliomas. One patient presented with multifocal disease, manifesting as a vagal PGL concurrent with bilateral carotid PGLs. Beyond multifocality, the SDHD H102R variant was also associated with tumor recurrence in one patient with a carotid PGL. None of the patients with metastatic disease were found to carry the SDHD H102R variant.
In the control cohort, the heterozygous SDHD H102R variant was identified at a frequency of 0.6% (6/1,055; 95% CI: 0.1–1.0%) among healthy Russian individuals. These variant carriers included both male and female subjects with a median age of 72.5 years. Comparing the variant frequencies between the case and control cohorts yielded an unadjusted odds ratio (OR) of 14.1 (95% CI: 5.4–37.3, p = 0.0001; Table 2). A chi-square test confirmed a highly significant association between the SDHD H102R variant and HNPGLs (p < 0.00001). After adjusting for cohort covariates such as sex and age, the adjusted OR remained strongly significant at 8.7 (95% CI: 2.3–32.6, p = 0.001; Supplementary Table 1).
The odds ratio estimates for the
| Parameter | Value | 95% Confidence Interval | |
|---|---|---|---|
| Unadjusted odds ratio (OR) | 14.1 | 5.4 – 37.3 | 0.0001 |
| Adjusted odds ratio (OR) | 8.7 | 2.3 – 32.6 | 0.001 |
The SDHD H102R variant demonstrated a maximum population frequency (combined data from all databases used) of 0.002% and a notable degree of evolutionary conservation (PhyloP score for primates = 8.7, phastCons score for primates = 1). The pathogenicity of the variant was predicted by all the in silico algorithms that were utilized.
Discussion
The heterozygous missense variant SDHD p.H102R was initially documented in a young woman with metastatic carotid PGL 18. This case provided the first evidence suggesting the potential pathogenicity of the SDHD H102R variant and its possible contribution to metastatic disease. More recently, Shulskaya et al. identified this variant in the peripheral blood of three Russian patients with multifocal HNPGLs 19. In our previous study, we reported the presence of the SDHD H102R variant in 9% of 134 Russian patients diagnosed with HNPGLs 12. In the current study, genetic testing of expanded case and ethnically matched control cohorts revealed the SDHD H102R variant in 7.5% of patients with HNPGLs and in 0.6% of healthy individuals. General population databases report a maximal allele frequency of 0.002% for this variant, which is significantly lower than the frequency observed in our Russian control cohort. It is hypothesized that the elevated frequency of the SDHD H102R variant in the Russian population may be attributable to a founder effect. Such founder SDHD mutations associated with pheochromocytoma and paraganglioma have been previously documented in the Netherlands 8 and China 20. Further investigation into this phenomenon is essential for improving diagnostics and clinical management within this population.
Notably, the six healthy individuals identified as carriers of the SDHD H102R variant were all over 70 years of age. In contrast, the median age of onset for SDHD H102R-related tumors is 50 years. Consequently, the likelihood of future disease manifestation in these elderly carriers is considered exceptionally low. The lack of penetrance in these cases may be explained by the parent-of-origin effect characteristic of SDHD-mutated hereditary PGLs. Because tumor development typically occurs exclusively when the SDHD mutation is paternally inherited (maternal imprinting) 21, it is highly probable that the SDHD H102R variant observed in these healthy subjects was maternally inherited.
According to the latest data from the ClinVar database (accessed October 2, 2025), this variant is currently classified as “pathogenic/likely pathogenic.” Based on the 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines for germline variant classification, the SDHD p.H102R variant was previously interpreted as “likely pathogenic” 22. The findings of the present case-control study indicate a strong association between the SDHD H102R variant and HNPGL, as evidenced by an adjusted odds ratio (aOR) of 8.7 (95% CI: 2.3–32.6, p = 0.001). Furthermore, multiple in silico algorithms predict the variant to be pathogenic. Consequently, these results fulfill additional ACMG/AMP 2015 criteria—namely, PS4 (based on the significantly elevated variant frequency in cases versus controls, as reflected by the aOR) and PP3 (based on supporting computational evidence).
In addition, the SDHD H102R variant is located within a recognized mutational hotspot, with other established pathogenic missense changes (p.His102Leu, p.His102Tyr, and p.His102Asn) occurring at the exact same amino acid residue. Collectively, these lines of evidence support the definitive classification of this variant as “pathogenic” within the Russian population. This genotype-phenotype association is further corroborated by existing submissions to the ClinVar database. Multiple submitters have reported the germline SDHD H102R variant in patients with a spectrum of related disorders, including pheochromocytoma, paraganglioma syndrome 1, hereditary cancer-predisposing syndrome, Carney-Stratakis syndrome, and Cowden syndrome 3. All of these pathologies are clinically associated with the development of paragangliomas, reinforcing the identified genotype-phenotype correlations.
A key limitation of this study is the disparity in the female-to-male ratio and age distribution between the case and control cohorts. To address this issue, a multiple logistic regression model was applied. This model demonstrated that sex differences did not significantly influence the analysis. Conversely, age was identified as a statistically significant predictor, exhibiting a negative correlation with HNPGL. Ultimately, both covariates were appropriately accounted for in the adjusted OR.
Furthermore, it is imperative to acknowledge potential survival and birth cohort biases introduced by the advanced median age of the control population (71 years). While the value of this older control group lies in demonstrating that SDHD H102R carriers can attain advanced ages without manifesting HNPGL—a critical point for genetic counseling regarding disease penetrance and age-related risk—it may not perfectly represent the variant's allele frequency in the general population at the typical age of disease onset. However, the impact of these biases is mitigated by several factors. First, heterozygous SDHD mutations are predominantly associated with paragangliomas and pheochromocytomas, which are rare neoplasms with generally low mortality rates. Existing evidence indicates that these mutations do not increase all-cause mortality 23,24. Consequently, a significant survival bias artificially altering the variant's frequency in elderly controls appears unlikely. Second, the strong genetic predisposition to HNPGL, combined with the absence of generationally variable environmental risk factors, minimizes the potential for birth cohort bias to influence the observed genetic association.
An additional limitation is the unknown germline versus somatic mutational status for 3 of the 14 patients harboring the variant. Unfortunately, the procurement of matched normal tissue samples from these patients for confirmatory genetic testing was not feasible. However, somatic coding SDHD mutations are exceptionally rare in paragangliomas and pheochromocytomas 25. Furthermore, an analysis of the variant allele frequency (VAF) in these cases revealed values approximating 0.5, strongly suggesting a germline origin. Consequently, it is highly probable that the variants of undetermined status are germline-derived.
Incomplete variant penetrance may have also influenced the analysis. As previously noted, SDHD mutations are subject to maternal imprinting. Therefore, investigating the transmission pattern of the SDHD H102R variant in the identified carriers remains a subject of great clinical interest. Finally, while our study established a robust statistical association between the variant and the disease, and in silico algorithms predicted a pathogenic effect, further functional studies are required to comprehensively elucidate its true pathogenicity.
Conclusion
The principal novelty of this study lies in the definitive validation and quantitative assessment of the association between the SDHD H102R variant and the risk of developing HNPGL. Utilizing a robust case-control design, we present key statistical evidence—namely, an odds ratio (OR) > 5.0 with a confidence interval excluding 1.0 (ACMG/AMP criterion PS4). When combined with other established lines of evidence, this strongly supports the classification of the SDHD H102R variant as “pathogenic” within the Russian population. These additional supporting criteria include the variant's low frequency in the general population (PM2), its localization within a recognized mutational hotspot containing several previously described pathogenic missense changes (PM1, PM5), pathogenic predictions by in silico algorithms (PP3), and its current “pathogenic” classification in the ClinVar database (PP5). However, it is important to note that our clinical and statistical findings do not preclude the need for future functional studies to definitively confirm the variant's underlying pathogenic mechanisms.
Abbreviations
ACMG/AMP: American College of Medical Genetics and Genomics and the Association for Molecular Pathology; aOR: adjusted odds ratio; CI: confidence interval; FFPE: formalin-fixed, paraffin-embedded; HNPGL: head and neck paraganglioma; OR: odds ratio; PCR: polymerase chain reaction; PGL: paraganglioma; SD: standard deviation; SDHx: succinate dehydrogenase; VAF: variant allele frequency.
Acknowledgments
We thank the Pathological Department, Vishnevsky Institute of Surgery for tumor sample collection and the Laboratory of Biological Microarrays, Engelhardt Institute of Molecular Biology for providing control samples. This work was performed using the equipment of the EIMB RAS “Genome” center ().
Author’s contributions
Conceptualization, A.S.; methodology, M.F., A.I., and M.E.; formal analysis, V.P.; resources, D.K.; writing - original draft preparation, A.S.; writing-review and editing, A.S. and A.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 24-14-00439.
Availability of data and materials
All data generated or analyzed during the study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Vishnevsky Institute of Surgery (Approval number 007/18) and was performed in accordance with the Declaration of Helsinki. Informed consent for the molecular genetic study and publication of the results was obtained from all patients involved in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.