
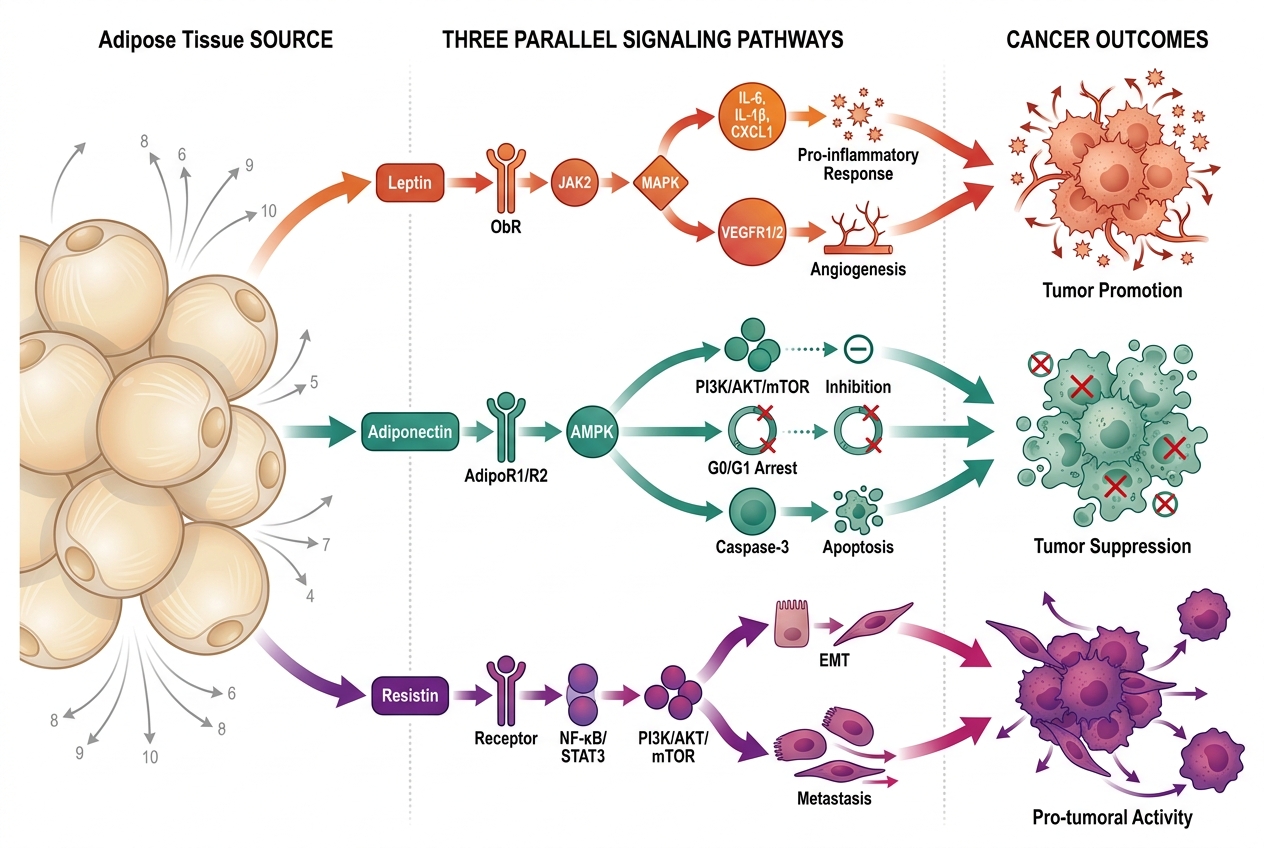

Adipokines as an inducer factor in the initiation and development of various types of cancer cells in the body
- Department of Biology, College of Sciences, University of Mosul, Iraq
Abstract
Hormones, including adipokines, are significant risk factors for the development of various cancers. Adipokines, also known as adipocytokines, are hormones synthesized by adipose tissue that play a crucial role in various metabolic and cellular processes. The aberrant expression of these hormones during obesity significantly contributes to chronic inflammation, insulin resistance, dysregulated lipid metabolism, cardiovascular disease, and cancer. This large group of cytokines interacts with multiple organs throughout the human body, including the breast, lungs, colon, and prostate gland. Leptin and resistin, two prominent adipokines, are particularly noteworthy for their role in activating cell survival and proliferative signaling pathways. Conversely, adiponectin overexpression exerts tumor-suppressive effects by inducing apoptosis and cell cycle arrest. In this review, we examine the role of adipocytokines in the growth and progression of malignancies across human and murine cancer models. Understanding the molecular mechanisms that mediate adipokine signaling in carcinogenesis is not only essential for fundamental research but could also lead to the development of novel therapeutic strategies for individuals at an elevated risk of cancer.
Introduction
Cancer remains the second leading cause of death worldwide. Global cancer statistics estimated 20 million new cases and 9.7 million deaths annually in 2022 1. Beyond established risk factors for carcinogenesis—such as family history, genetic mutations, ionizing radiation, chemical exposures, infectious agents, poor diet, alcohol and tobacco consumption, and a sedentary lifestyle—obesity remains a primary risk factor for various malignancies. This is due to its correlation with other systemic factors, such as chronic inflammation, hormonal disorders, impaired immune function, and metabolic alterations. These systemic changes significantly contribute to the induction of a microenvironment that promotes the growth and progression of cancerous cells 2,3,4.
Obesity is not merely the accumulation of excessive fat deposits causing health problems such as insulin resistance and cardiovascular disease; it is also a leading risk factor associated with the progression of 13 diverse types of malignant cancers and an increased cancer-related mortality rate of up to 17% 4,5. The accumulation of large amounts of carbohydrates, proteins, and lipids in adipose tissue promotes the release of inflammatory mediators, such as prostaglandins, cytokines, and chemokines, which contribute to tumorigenesis 6,7. Numerous studies have demonstrated that obesity is correlated with risk factors for the development of several cancers 8.
Adipokines are polypeptides and bioactive mediators primarily produced by adipose cells. This active endocrine organ synthesizes and secretes more than 50 cytokines and hormones, maintaining homeostasis under normal conditions 9. The dysregulation of adipokines can lead to inflammation and diseases such as insulin resistance, cardiovascular disease, and atherosclerosis, while also affecting obesity, energy homeostasis, immune responses, and metabolic activities 9,10,11. Adipokines include adiponectin, leptin, resistin, vaspin, apelin, visfatin, chemerin, and omentin; their type and secretion levels depend on the size, location, and number of adipocytes in the tissue 12,13.
Epidemiological studies have found that an increase in circulating adipokines—such as leptin, resistin, visfatin, osteopontin, apelin, and lipocalin—plays a prominent role in carcinogenesis through multiple mechanisms, leading to cancer cell proliferation. Conversely, low levels of specific circulating adipokines have been associated with an increased risk of cancer formation 14. Furthermore, these adipokines mediate metastasis and angiogenesis by interacting with components of the tumor microenvironment across various cancer types 15.
Function of Leptin in Cancerogenesis
Leptin, a 16 kDa protein encoded by the obesity gene (ob) located on chromosome 7q32.1, was identified by Dr. Jeffrey Friedman and his team in 1994 as a multifunctional hormone with a wide range of physiological roles 16. Adipose tissue is the primary source of leptin production; however, many other organs and tissues also express it, including the small intestine, brain, liver, ovaries, and skeletal muscle 15,17.
Leptin regulates body weight and energy balance by suppressing appetite, which leads to reduced adipocyte lipid storage and a decrease in overall body fat. Furthermore, it regulates bone formation, fetal growth, reproduction, and milk production in the mammary gland 18. As a key adipocytokine involved in preventing obesity 19, leptin deficiency or leptin receptor dysfunction can lead to increased food intake, contributing to human obesity and the induction of cancer 20.
Leptin influences tumor-associated activities by promoting various signaling pathways, manipulating the tumor microenvironment, and interacting with other molecules to stimulate carcinogenesis. It plays a critical role in regulating molecules involved in cell proliferation, differentiation, metastasis, angiogenesis, and inflammatory processes that support tumor development (Figure 1). Consequently, targeting the leptin signaling pathway could pave the way for innovative treatments for obesity-related cancers 21.
Research indicates that leptin significantly contributes to the initiation and development of various types of cancer, particularly colon cancer. One study showed that serum leptin concentrations in patients with poorly differentiated colon tumors were dramatically lower than in healthy individuals 22. In another study, leptin reduced the development and growth of colon cancer in murine models (in vivo). In contrast, it promoted the growth of HT-29 colon cancer cell lines in vitro by stimulating DNA synthesis and cellular proliferation 23.
Furthermore, elevated leptin levels have been shown to increase colon epithelial cell proliferation and inhibit apoptosis, linking obesity and colon cancer 24. Elevated leptin levels promote colon tumor growth in obese patients by inducing inflammatory mediators associated with colon cancer, such as IL-6, IL-1β, IL-1, and CXCL1. Additionally, it promotes cell proliferation by upregulating the transcription of Notch-4, Notch-1, JAK-2, STAT1, and STAT3. Moreover, high leptin expression can induce angiogenesis via VEGFR1 and VEGFR2 in several cancer types 25,26.
Leptin reduces apoptosis by binding to its specific cell surface receptor (Ob-R), which stimulates the PI3K/Akt and MAPK/ERK1/2 signaling pathways. These pathways play vital roles in inducing cell proliferation and inhibiting apoptosis in three human colon cancer cell lines (T84, HT29/C1.19A, and Caco-2) 27. Moreover, experiments using tumor tissues and cell lines suggest that decreased levels of leptin induce tumor necrosis factor-alpha (TNF-α), which promotes apoptosis. The molecular mechanism of TNF-α-induced cell death involves upregulation of p53-independent cell death through increased expression of the p53-upregulated modulator of apoptosis (PUMA) under stressful conditions 28. A meta-analysis of twenty-three studies has shown a strong correlation between elevated circulating leptin levels and colorectal cancer risk 29.
In breast cancer, leptin acts as a growth factor similar to insulin, promoting cell growth, invasion, and angiogenesis by binding to the leptin receptor (ObR) and activating the PI3K/Akt/mTOR and JAK/STAT3 pathways 30,31. A 2012 study found that the oncoprotein human epidermal growth factor receptor 2 (HER2) significantly induced transcription of the leptin gene, thereby augmenting leptin expression by approximately 30% in breast cancer via the p38 mitogen-activated protein kinase (MAPK) signaling pathway. In turn, leptin regulates the overexpression of MMP2 and MMP9, which are involved in tumor cell invasion in MCF10A breast cancer cells 32. Additionally, leptin appears crucial for promoting migration and invasion in the MCF10A breast cancer cell line (in vitro) by upregulating kinases such as Src and FAK, which regulate the expression of Twist and β-catenin. Twist and β-catenin are transcription factors that bind the promoters of the MMP2 and MMP9 genes to promote their transcription 33.
Furthermore, several researchers have found an association between high levels of leptin and prostate cancer. The mechanisms by which leptin induces cell proliferation in androgen-independent DU145 and PC-3 cells involve the activation of JNK. In contrast, leptin did not influence cell proliferation in androgen-dependent prostate cancer LNCaP-FGC cells 34. Other studies have shown that leptin increases cell proliferation in androgen-independent DU145 cells by upregulating the phosphorylation of MAPK, ERK1/2, and the PI3K/Akt signaling pathways 35,36.
In lung cancer, leptin promotes metastasis by activating the ERK and AKT pathways, which play a crucial role in modulating epithelial-mesenchymal transition (EMT) 37. Autocrine leptin produced by lung cancer cells can promote cancer cell growth in non-small cell lung cancer (NSCLC) patients. Overexpression of leptin receptors promotes the initiation and progression of malignancy in two human NSCLC cell lines, H1299 and A549. It positively regulates the PI3K/AKT/mTOR signaling pathway and negatively regulates the PI3K/AKT/MDM2/P53 signaling pathway, both of which are associated with cancer proliferation, invasion, and metastasis. Furthermore, leptin can induce cell cycle arrest in the lung cancer cell line A549-NC by upregulating Cyclin D1 and CDK2 genes and downregulating p53, which are essential proteins that regulate the G1/S checkpoint. Leptin also induces autophagy by increasing LC3-II levels and decreasing P62 expression in the H1299 lung cell line 38.
Function of Adiponectin in Cancerogenesis
Adiponectin, a bioactive protein that exerts its effects primarily through the AdipoR1 and AdipoR2 receptors, is an abundantly produced hormone first identified in 3T3-L1 adipocytes and found at high levels in murine plasma 39. Scherer and colleagues were the first to identify adiponectin from the murine adipocyte cell line (3T3-L1), naming it Acrp30 (adipocyte complement-related protein of 30 kDa). It acts as a multifunctional molecule with anti-inflammatory, anti-atherogenic, antioxidant, and antiproliferative properties. Recognized as a regulator of glucose homeostasis and lipid metabolism in muscle and liver cells, adiponectin is often colloquially referred to as a "guardian angel" of metabolism. Its crucial role in regulating insulin levels and lipid metabolism has inspired the development of novel therapies for type 2 diabetes mellitus, other insulin-resistant conditions, cardiovascular disease, and various cancers 40,41.
Mechanistically, the AdipoR1 receptor stimulates AMP-activated protein kinase (AMPK), which governs cellular energy homeostasis and phosphate metabolic activities. Concurrently, AdipoR2 controls the expression of the peroxisome proliferator-activated receptor α (PPARα) gene, a critical regulator of human steroidogenesis, fatty acid oxidation, and anti-inflammatory responses 42,43.
Conditions such as obesity, type 2 diabetes, atherosclerosis, cardiovascular diseases, and hypertension are associated with decreased adiponectin production by adipocytes. Obesity induces significant adipokine dysregulation, leading to the upregulation of oncogenic leptin and the downregulation of tumor-suppressive adiponectin; this imbalance can drive cancer development in various organs 41,44. Additionally, obesity reduces the activity of AdipoR1/R2, leading to adiponectin resistance—a state wherein cells become less responsive to the hormone. In this condition, adiponectin cannot effectively bind to its receptors or activate downstream intracellular pathways, even in the presence of normal or elevated circulating adiponectin levels. This phenomenon is attributed to the overexpression of APPL2, which binds to the adiponectin receptor and inhibits downstream signaling pathways. Such resistance precipitates several metabolic disorders, including insulin resistance, type 2 diabetes, and cardiovascular disease, and is intimately linked to obesity-induced inflammation 45.
Classical adiponectin receptor expression is significantly downregulated in diabetes and obesity, resulting in the suppression of AMPK activation, which normally regulates glucose uptake and lipid oxidation for energy production in muscle and liver tissues. The receptor's action is directly associated with insulin levels; notably, adiponectin receptor expression increases markedly in the skeletal muscle and adipose tissue of mice during fasting, whereas systemic insulin levels rise after refeeding 46,45.
In oncology, adiponectin has been shown to play a protective role in tumor initiation and progression by regulating multiple signaling pathways that restrict cell growth, stimulate apoptosis, and inhibit angiogenesis (Figure 1) 15. Researchers have observed, using animal models and human cancer cells, that adiponectin receptor expression and circulating adiponectin levels could offer therapeutic targets and serve as biomarkers for cancer diagnosis and prognosis 47. The depletion of the adiponectin receptors AdipoR1 and AdipoR2 has been observed in various cancer cell lines. Glioblastoma, breast cancer, head and neck squamous cell carcinoma, liver cancer, lung cancer, and renal cell carcinoma exhibit dysregulation of AdipoR1, while AdipoR2 protein levels are commonly downregulated in breast and liver cancers 48.
Recent data indicate that adiponectin and its receptor expression levels are crucial factors in the development of prostate cancer. Serum adiponectin levels are notably reduced in prostate cancer patients compared to healthy individuals, and the administration of synthetic adiponectin has demonstrated therapeutic potential in protecting against cancer occurrence in animal models. Consequently, this suggests a potential role for adiponectin in suppressing cell proliferation and inducing apoptosis in prostate cancer 49. In prostate cancer cells, the reduction of adiponectin inhibits the AMPK/TSC2 signaling pathway. Conversely, adiponectin activation downregulates mTOR expression, which suppresses vascular endothelial growth factor-A (VEGF-A) activity and inhibits angiogenesis in PC-3 prostate cancer cells 50. Furthermore, adiponectin induces cell cycle arrest at the G0/G1 phase in prostate cells by downregulating cyclin D1 and proliferating cell nuclear antigen (PCNA). It also stimulates apoptosis via the activation of caspase-3 and the MEK/ERK/p90RSK signaling pathway in RWPE1 and WPMY1 prostate cancer cells 51. Kashiwagi reported that exogenous adiponectin suppresses migration by inhibiting the expression of NF-kB, STAT3, and COX-2 transcription factors, which are drivers of cell migration in several prostate cancer cell lines, including PC-3, LNCaP, 22Rv1, and DU145 52.
The role of adiponectin in breast cancer cell proliferation is highly dependent on the estrogen receptor subtype (ERα-positive vs. ERα-negative). Many epidemiological studies have documented that adiponectin and its receptors act as protective factors against the initiation and development of ER-negative breast neoplasms. Potential mechanisms by which adiponectin suppresses cell growth and induces cell cycle arrest in ER-negative breast cancer involve decreased expression of cyclin D1 (CCND1). This occurs through the phosphorylation of AMPK, which in turn increases the phosphorylation of SP1, a transcription factor for the CCND1 promoter. This cascade restricts the movement of RNA polymerase II and promotes the attachment of a corepressor complex containing HDAC1, SMRT, and NCoR 53. Several studies have well-documented the role of adiponectin in modulating numerous intracellular signaling pathways, primarily by activating the dominant AMPK pathway after binding to its receptors (AdipoR1 and AdipoR2). In turn, AMPK inhibits the PI3K/AKT signaling pathway, thereby inhibiting mTOR; this is significant because the PI3K/AKT/mTOR pathway is heavily implicated in breast cancer cell growth and migration 54,55. Additionally, AMPK suppresses the activity of c-Jun N-terminal kinase (JNK), signal transducer and activator of transcription 3 (STAT3), and the NF-κB signaling pathway. Simultaneously, AdipoR2 inhibits ceramide activity, which has been linked to insulin resistance, apoptosis, inflammation, and atherosclerosis 55.
Conversely, when ERα-positive breast cancer cells (such as MCF-7) are treated with exogenous adiponectin, it can increase cyclin D1 expression by phosphorylating SP1 and ERα. This enhances the recruitment of RNA polymerase to the promoter, boosting cyclin D1 production and ultimately increasing breast cancer cell proliferation 56. A recent study has shown that adiponectin can inhibit breast cancer proliferation by downregulating fatty acid synthesis (FAS) via the activity of SIRT1. SIRT1 plays an essential role in adiponectin's metabolic effects within breast cancer cells, as it can either suppress SREBP1-mediated transcription of FAS genes or stimulate autophagy-promoted lipolysis and fatty acid oxidation (FAO). Both mechanisms deplete the cellular fatty acid pool, ultimately leading to the promotion of apoptosis in breast neoplasms. This study provides a novel mechanism for suppressing breast cancer cell growth 53.
Lung cancer is also associated with obesity; a positive correlation between obesity and lung cancer incidence has been well-established 57. Furthermore, serum adiponectin levels decrease with the development of lung cancer. Paradoxically, the expression of AdipoR1 at both the gene and protein levels is higher in patients with lung cancer than in healthy individuals, while AdipoR2 is only slightly increased in both groups 58. Adiponectin has demonstrated a role in reducing cell proliferation and survival by inducing an increase in lipid peroxidation in human lung adenocarcinoma A549 cells 58. Adiponectin possesses an anti-migration effect by increasing the expression of E-cadherin and lowering vimentin expression—key markers involved in cellular attachment and epithelial-mesenchymal transition (EMT)—in three non-small cell lung carcinoma cell lines (NCI-H1299, HCC827, and A549) 59.
Adiponectin exerts its effects via several intracellular signaling pathways. Regarding colon cancer—one of the most common human malignancies—numerous studies have indicated that a low level of adiponectin is a significant risk factor for its development 60. Dysregulation of adiponectin and its receptors plays a crucial role in the growth and progression of colon tumors 61. In obesity-promoted inflammation, hypoxic adipocytes secrete factors that promote macrophage recruitment. A low adiponectin level in adipose tissue exacerbates the production of tumor necrosis factor-alpha (TNF-α) and monocyte chemotactic protein-1 (MCP-1). Specifically, these inflammatory mediators can activate the Toll-like receptor 4 (TLR4) and its downstream NF-κB signaling pathway, thereby stimulating cancer growth 62. Fenton and Birmingham found that MC-38 colon cancer cells treated with adiponectin exhibited suppressed IL-6-induced cell proliferation via the downregulation of STAT3 activation, although it had no effect on insulin-induced cell proliferation. Furthermore, MC-38 cells treated with a combination of IL-6 and adiponectin demonstrated inhibition of IL-6-induced nitric oxide synthase (NOS), which plays a role in the progression of colon carcinogenesis 63. Paradoxically, it has also been observed that adiponectin can induce angiogenesis in certain colon cancer models.
The Function of Resistin in Carcinogenesis
Resistin is a small circulating protein initially discovered as an adipocyte-derived cytokine in mice 6. It is also referred to as C/EBP-epsilon-regulated myeloid-specific secreted cysteine-rich protein (XCP1) or adipose tissue-specific secretory factor (ADSF) (136). The primary function of resistin is to regulate glucose homeostasis in mammals. Consequently, high circulating resistin levels are associated with pathologies such as insulin resistance, cancer, obesity, cardiovascular disease, autoimmune diseases, and inflammatory states 64,62. The term resistin was initially coined by Dr. Steppan and colleagues from the University of Pennsylvania in 2001, reflecting its ability to induce insulin resistance in rodents, which in turn causes type 2 diabetes mellitus 65.
In human physiology, it has been reported that resistin is involved in inflammation, chaperone function, stress biology, and serves as a biological marker linking obesity and diabetes 66. High resistin concentrations have also been identified in pulmonary inflammation among patients with asthma 12. The primary sources of resistin secretion are white adipocytes in mature mice, whereas monocytes and macrophages are the central sites of resistin production in human adipocytes 66.
Resistin is divided into three family members based on their location, structure, and function: RELMα (Resistin-like molecule alpha), RELMβ (Resistin-like molecule beta), and RELMγ (Resistin-like molecule gamma) 62. The expression of RELMs is associated with several signaling pathways, including PI3K/Akt/mTOR, IL-4/IL-4Rα, and HMGB1/RAGE, which are responsible for many physiological processes and pathogenesis in mammals, such as cancer 68,69.
RELMα, a small peptide rich in cysteine, is primarily secreted in white adipose tissue, especially in murine gonadal fat, while it is present in the lung, heart, and tongue in humans 70. Research has also demonstrated that RELMα is secreted by skin cells and acts as a protective factor against Streptococcus pyogenes infections in the skin in vivo, with vitamin A being essential for its expression 71. Other studies have localized RELMα in dendritic cells, macrophages, type II alveolar epithelial cells (AEC II), and pulmonary microvascular endothelial cells (PMVECs), where it plays a role in lung diseases such as pulmonary inflammation, asthma, and fibrosis, and induces pulmonary hypertension 72,73. Additionally, data indicate that high expression of RELMα was significantly associated with the promotion of growth and progression in the gastric cancer cell lines SGC7901 and MKN45 via the activation of nuclear factor-kappa B (NF-κB), vascular endothelial growth factor (VEGF), and MMP-9. At the same time, inhibition of RELMα activity via Ad5/F35-siRNA treatment significantly reduced NF-κB, VEGF, and MMP-9 expression, which are involved in migration and angiogenesis 74.
RELMβ, also known as FIZZ2, is expressed in the epithelial cells of the gastrointestinal tract and stimulates the growth of colon cells 75. It has also been found to induce several lung diseases in humans, such as asthma, fibrosis, inflammation, and remodeling, by promoting hypoxia-induced proliferation of lung cells 76. RELMβ has a crucial, albeit complex, role in carcinogenesis. The overexpression of RELMβ can facilitate invasion and metastasis in the AGS gastric cell line by activating N-cadherin, Snail, and vimentin while inhibiting E-cadherin levels, thereby promoting epithelial-mesenchymal transition (EMT) 77. In contrast, another study found that high expression of RELMβ suppressed the migration and invasion of the gastric cancer cell lines SGC-7901 and MKN-45 by downregulating MMP-2 and MMP-9 expression. Furthermore, RELMβ can abolish the expression of VEGF, which induces angiogenesis in cancer cells; this study suggested its potential utility as a novel therapeutic approach for gastric cancer 78. In colorectal cancer (CRC), the expression of RELMβ was measured in CRC biopsies; the data showed a significant reduction in the level of RELMβ in CRC patients harboring mutations in KRAS, TP53, and BRAF compared to healthy individuals 79. RELMγ is expressed in white blood cells, the spleen, the thymus, and the nasal respiratory epithelium of cigarette-smoked rats 62.
Four distinct cell-surface receptors for resistin have been identified, including adenylyl cyclase-associated protein 1 (CAP1), an isoform of decorin (ΔDCN), receptor tyrosine kinase-like orphan receptor 1 (ROR1), and Toll-like receptor 4 (TLR4), all of which trigger the activation of various signaling pathways within cells 62. The interaction of resistin with CAP1 increases the activation of the cAMP signaling pathway in monocytes, induces protein kinase A (PKA) activity, and promotes NF-κB-mediated transcription of inflammatory cytokines (IL-6, TNF-α, and IL-1β) 80. Another study has shown that CAP1 is involved in insulin resistance, inflammation, and cell death in murine hepatocytes (159). The binding of TLR4 to resistin upregulates inflammatory effects, and in lung cells, promotes chronic obstructive pulmonary disease (COPD) through autophagy 81. The binding of resistin to decorin promotes cell proliferation and metastasis, and the decreased production of lipids in the 3T3-L1 murine cell line results in the activation of extracellular signal-regulated kinases 1 and 2 (ERK 1/2) 81. The interaction of resistin with ROR1 significantly regulates glucose metabolism and adipocyte differentiation by modulating GLUT1 and GLUT4 expression. Also, the overexpression of ROR1 upregulates the phosphorylation of ERK1/2, AKT (Thr308), GSK3β, and AMPK, and decreases AKT (Ser473) and p38 MAPK phosphorylation in murine cell lines 83.
Aberrant expression of resistin and its receptors is correlated with many types of malignancies (Figure 1). Several studies have demonstrated that high expression of resistin, through its varied receptors, promotes cell growth, invasion, migration, angiogenesis, inflammation, and drug resistance 6,62. Epidemiological studies have shown that increased plasma resistin activity upregulates prostate tumor cell secretion of extracellular vesicles. These lipid-bound vesicles mediate various stages of tumor initiation and progression, including cell proliferation, angiogenesis, migration, and invasion. Furthermore, resistin increased MMP-2 and MMP-9 release and induced cell metastasis by activating p-FAK in resistin-treated PC3 cells 84. Another study on prostate cancer found that resistin increases the mRNA production of suppressors of cytokine signaling (SOCS3 and SOCS5). This upregulation of SOCS3 and SOCS5 expression plays a crucial role in increased cell proliferation in PC-3 human prostate cancer cells by correlating with resistin and TLR4 and activating signaling pathways such as PI3K/AKT, p38/MAPK, and ERK 85.
In breast cancer, elevated circulating resistin concentrations increased cellular motility and cell migration in the MCF-7 and MDA-MB-231 cell lines. The interaction of resistin with the receptor CAP1 activates the signal transduction pathway. It induces the expression of proteins SNAIL, SLUG, ZEB1, TWIST1, fibronectin, and vimentin during epithelial-mesenchymal transition (EMT), and reduces the expression of epithelial markers (E-cadherin and claudin-1). Thus, resistin significantly contributes to breast cancer cell metastasis 86. Furthermore, other studies have indicated that resistin, acting through its receptor Toll-like receptor 4 (TLR4), is a risk factor for breast cancer initiation and progression by activating the NF-κB/STAT3 signaling pathway 87.
In colon cancer cells, resistin overexpression correlates with cell cycle arrest. Binding of resistin to the TLR4 receptor and the subsequent active TLR4-MyD88 interaction causes the phosphorylation of ERK, which in turn induces the expression of SOCS3. An increase in SOCS3 expression activates the JAK2/STAT3 pathway, resulting in the arrest of HCT116 colon cancer cells in the G1 phase 88. Additionally, a significant reduction in RETNLB expression was observed in colon cancer cells and was associated with mutations in the p53, BRAF, and KRAS genes 79. A high amount of resistin has also promoted colon cancer cell metastasis by activating NF-κB, which regulates the intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). ICAM-1 and VCAM-1 can facilitate cancer cell adhesion to the endothelium and the migration of malignant cells 89.
Regarding lung cancer, resistin plays a notable role in its development. Resistin levels were higher in cachexia patients with non-small cell lung cancer (NSCLC) and are associated with cancer through its vital role as a proinflammatory cytokine 90. A recent study demonstrated significantly increased resistin levels in patients with NSCLC compared with healthy people in Iraq 89. Excessive expression of resistin on Toll-like receptor 4 (TLR4) also promoted the migration and invasion of A549 lung adenocarcinoma cells. The interaction of resistin with TLR4 significantly contributes to increased phosphorylation of the Src and EGFR signaling pathways, which upregulates the expression of PI3K and NF-κB. NF-κB subsequently acts as a transcription factor for the MMP2 and TWIST1 genes, inducing migration and invasion 90.
Summary of the mechanism pathways of adipokines on different type cancer cells in the body.
| Cancer Type | Adipokine Type | Actions | Physiological state |
|---|---|---|---|
| endometrial cancer | Leptin ↑ |
Activate the expression of tumorigenic proteins, including IGF-1 and VEGF Induce JAK/STAT and MAPK-ERK pathways |
Angiogenesis Cell proliferation |
| Adiponectin ↓ | Decrease the expression of activated, phosphorylated-AMPK (p-AMPK) and increased phosphorylation of mTOR (p-mTOR) | Apoptosis | |
| Resistin ↑ | Activate PI3K/Akt pathway. | Cell proliferation | |
| Breast cancer | Leptin ↑ |
mitogen-activated protein kinase (MAPK) and STAT3 signaling cascades. HIF1α and PKM2 induced angiogenesis Necrosis factor α (TNFα) and IL-6 |
Migration Angiogenesis |
| Adiponectin ↑ |
Activating AMKP and inhibiting MAKP, Inhibition of Cyclin D1 through the Wnt pathway ,Activating LKB1 and PP2A, Activating Bax, Caspase 8, and P53 |
Cell proliferation Cell cycle arrest Invasion Apoptosis | |
| Resistin ↑ |
Upregulation of CSC markers SOX2, NANOG, and OCT4 Silencing adenylyl cyclase-associated protein 1 (CAP1) | Invasion | |
| Prostate cancer | Adiponectin↑ |
Activate AMPK-induced tuberous sclerosis protein 2 (TSC2), causing inhibition of rapamycin (mTOR), vascular endothelial growth factor A (VEGF-A) Activation of Signal transducers and activator of transcription 3 (STAT3) activity Increasing caspase 3, Bax expression, P53, and downregulation of Bcl2 |
Apoptosis Angiogenesis Apoptosis |
| Leptin ↑ | Activation JAK2/PI3K Activate SATA3signaling pathway | Cell proliferation | |
| Resistin ↑ | p-FAK levels and increased secretion of MMP-2 and MMP-9 in | Migration and invasion | |
| Colon cancer | Leptin ↑ | Stimulate MAPK/ ERK1/2, JAK2/SATA3 signaling pathway | Cell proliferation |
| Adiponectin ↑ | Activate (AMPK) pathway, while inhibiting the PI3K/AKT, mTOR, | Cell proliferation | |
| Resistin ↑ |
Simulates the production of proinflammatory cytokines such as TNF-α and IL-12, which are mediated by the NF-κB | Inflammation caused the growth cell | |
| Lung cancer | Leptin ↑ | Activate angiogenic factors such as (VEGF) and (FGF). | Angiogenesis |
| Adiponectin ↑ |
The inhibition of the nuclear factor-kappa B (NF-κB) signaling pathway Suppress matrix metalloproteinase 12 (MMP-12) | Cell Proliferation | |
| Resistin ↑ | Activating a signaling pathway involving Toll-like receptor 4 (TLR4), Src, epidermal growth factor receptor (EGFR), PI3K, and NF-κB. | Metastasis | |
| Ovarian cancer | Leptin ↑ |
Increase signaling via the JAK/STAT3 pathway upregulates ERα transcriptional activation Reduce the cytotoxic effect of PTX/TXT Enhanced expression MMP9-dependent manner |
Progression of cancer Migration |
| Adiponectin ↑ |
Enhances phosphorylation of AMPK. | Apoptosis | |
| Resistin ↑ |
Activate PI3K/Akt/Sp1 pathway Stimulate the mTOR pathway, | Cell proliferation | |
| Liver cancer | Leptin ↑ | activation of STAT3, ERK1/2, and Akt | |
| Adiponectin ↑ | Stimulate LKB/AMPK signaling pathway | Cell proliferation | |
| Resistin ↑ | Promote mitogen-activated protein kinases (MAPKs) and extracellular signal-regulated kinases (ERK). and MMP | Cell Proliferation Migration | |
| Brain cancer | Leptin ↑ | Activation of the Src/ERK/Akt/mTOR /p70S6K/rS6 pathway | Cell proliferation |
| Adiponectin ↑ |
Decreasing Cyclin D1 and Cyclin B1. Inhibit Akt/mTOR pathway |
Cell cycle arrest Cell proliferation | |
| Resistin ↑ | activate pathways like PI3K/Akt and p38 MAPK | Cell proliferation |
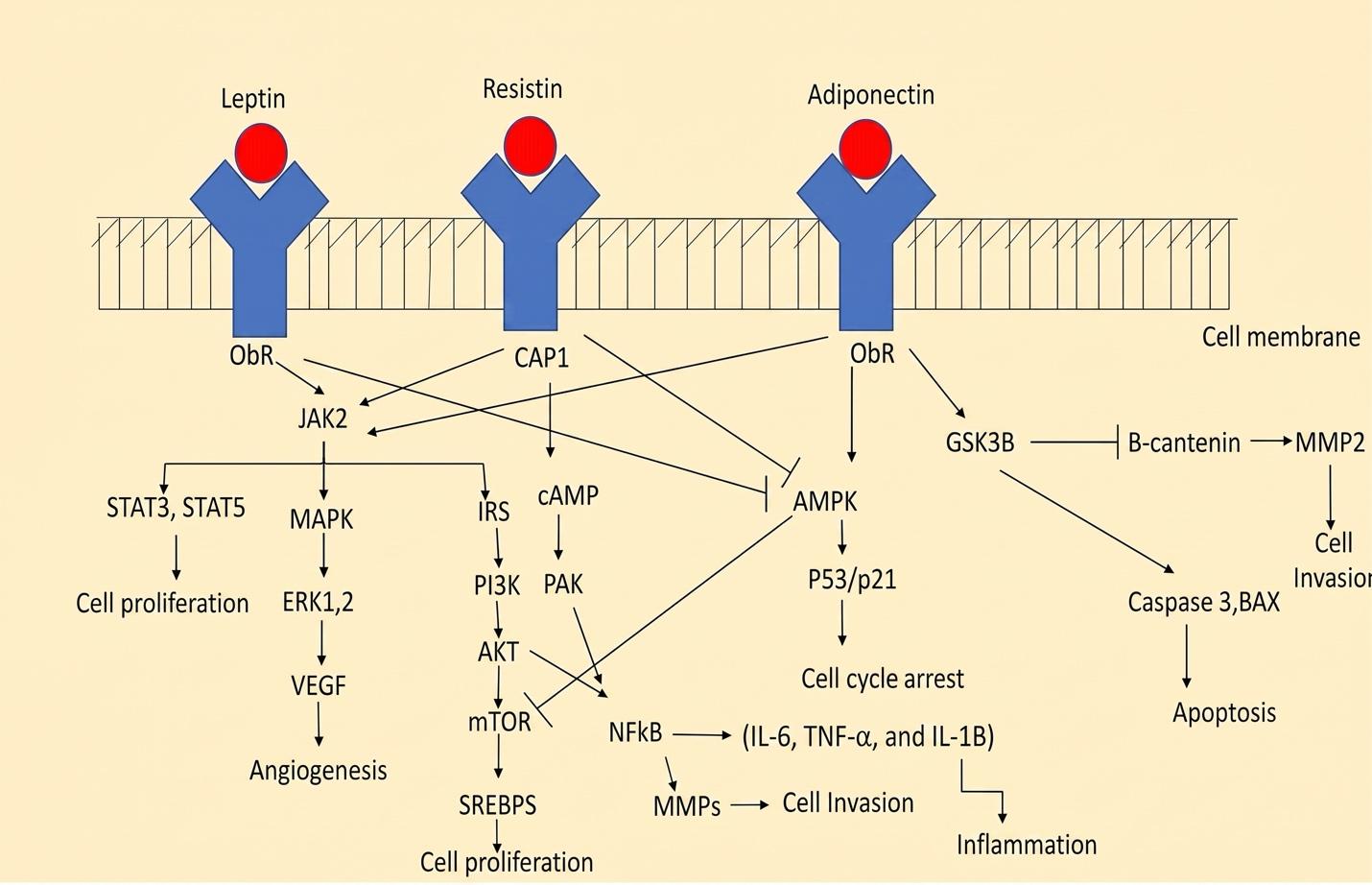
A schematic showing different types of adiponectin -induced various signaling pathways. The binding of leptin to its receptor (ObR) leads to the formation of the ObR/JAK2 complex, resulting in phosphorylation (P of the ObR). This phosphorylation activates MAPK/ERK1/2 signaling, PI3K/Akt, and downstream signals, including mTOR. Also, phosphorylated STAT3 and STAT5 translocated to the nucleus, where they activate target genes. Additionally, The attaching of resistin to its receptor (CAP1) triggers the formation of the ObR/JAK2 complex, which lead to activate MAPK/IRS, and SATA3 signaling, as well as the cAMP/PAK signaling pathway, and inhibit AMPK induced p53 ,While reaction Adiponectin with obR induce AMPK induce of cell cycle arrest, GSK38 induced of apoptosis.
Conclusion
In conclusion, obesity is a well-established risk factor for various malignancies. Scientific research on adipokines has elucidated the mechanistic link between carcinogenesis and obesity. These bioactive molecules are implicated in the pathogenesis of several hormone-related malignancies by modulating various mechanisms that drive cell proliferation, differentiation, migration, and angiogenesis across multiple cancer cell types. Broadly, a decline in adiponectin concentrations enhances the proliferation of cancer cells, while high concentrations of leptin and resistin contribute to an elevated risk of cancer. Understanding the molecular mechanisms of adipokines and establishing their clinical utility as biomarkers for cancer prevention and treatment may provide valuable insights for developing novel therapeutic interventions in the future.
Abbreviations
AMPK: AMP-activated protein kinase, EMT: Epithelial-mesenchymal transition, ERK: Extracellular signal-regulated kinase, MAPK: Mitogen-activated protein kinase, mTOR: Mammalian target of rapamycin, NSCLC: Non-small cell lung cancer, PI3K: Phosphoinositide 3-kinase, STAT: Signal transducer and activator of transcription, VEGF: Vascular endothelial growth factor
Acknowledgments
None.
Author’s contributions
FHK, AHA, and SZK conceptualized the review, conducted the literature search, and drafted the manuscript. All authors read and approved the final manuscript.
Funding
The authors declare that no specific funding was received for this study.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have used generative AI and/or AI-assisted technologies in the writing process before submission, but only to improve the language and readability of their paper.
Competing interests
The authors declare that they have no competing interests.