
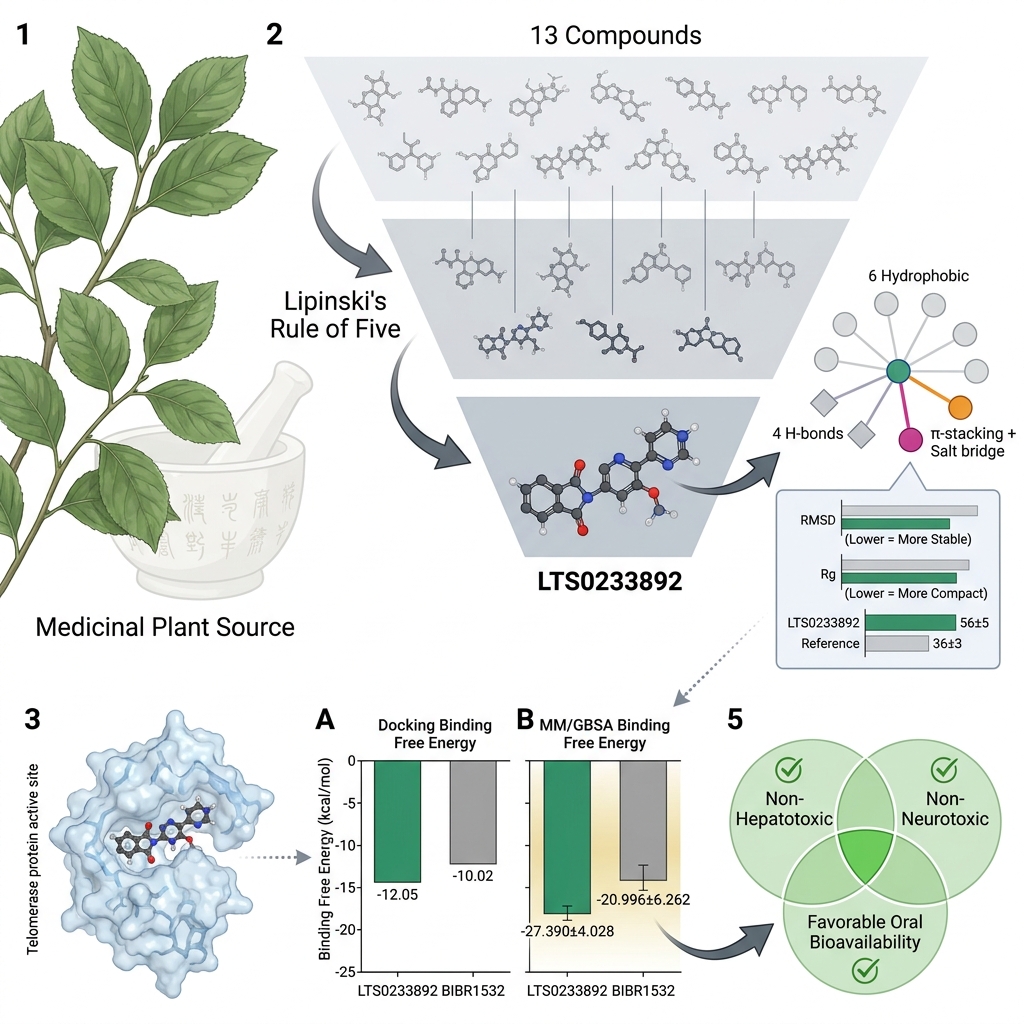

In silico identification of a stable and drug-like Telomerase inhibitor originating from Celastrus hindsii
- Graduate University of Science and Technology, Vietnam Academy of Science and Technology, Viet Nam
- Faculty of Natural science education, Saigon University, 273 An Duong Vuong, Cho Quan Ward, Ho Chi Minh City, Viet Nam
Abstract
Introduction: Telomerase is reactivated in approximately 90% of human malignancies, underscoring its potential as a cancer-selective therapeutic target with minimal off-target toxicity. Natural products derived from medicinal plants represent a promising and underexplored source of novel telomerase inhibitors. Despite its longstanding empirical use in Vietnamese traditional medicine, Celastrus hindsii remains insufficiently characterized at the molecular level.
Methods: In this study, we employed an integrated in silico framework to systematically evaluate C. hindsii-derived compounds for their telomerase-inhibitory potential.
Results: Among the screened candidates, LTS0233892 emerged as the leading compound, exhibiting enhanced binding affinity and sustained interactions within the enzyme's catalytic site. Molecular dynamics simulations over 200 ns revealed that the telomerase-LTS0233892 complex maintained greater structural stability than the complex formed with the reference inhibitor, demonstrating an average backbone root mean square deviation (RMSD) of 0.348 ± 0.042 nm compared to 0.425 ± 0.068 nm for BIBR1532. Consistently, radius of gyration analysis indicated a more compact protein conformation upon LTS0233892 binding, yielding an average value of 2.932 ± 0.029 nm, compared to a slightly higher value of 2.972 ± 0.026 nm for the BIBR1532-bound system. Mechanistically, principal component analysis combined with free energy landscape reconstruction demonstrated that LTS0233892 stabilizes a more energetically favorable conformational ensemble of telomerase compared to BIBR1532. These results are consistent with MM/GBSA binding free energy calculations, in which LTS0233892 exhibited a binding free energy of –27.390 ± 4.028 kcal/mol, whereas BIBR1532 demonstrated a weaker binding energy of –20.996 ± 6.262 kcal/mol. Furthermore, LTS0233892 exhibited favorable drug-like properties, including optimal aqueous solubility and predicted oral bioavailability.
Conclusion: Collectively, these computational findings indicate that LTS0233892 represents a promising small-molecule candidate warranting further experimental validation of its telomerase-inhibitory activity.
INTRODUCTION
In eukaryotes, the intrinsic limitations of DNA polymerases prevent the complete replication of the terminal ends of linear chromosomes during genome duplication. Consequently, each replication cycle results in progressive DNA shortening due to the removal of the RNA primers at the chromosome ends1,2. Telomeres function as specialized nucleoprotein assemblies that preserve chromosome integrity by shielding terminal regions from degradation and illegitimate recombination3. They typically comprise approximately 1,000 – 2,000 tandem repeats of a short DNA motif that adopts a T-loop configuration, reinforced by a protruding 3′ guanine-enriched single-stranded extension to ensure end stability4,5,6. Progressive telomere attrition to a critical threshold subsequently activates DNA damage signaling pathways, culminating in apoptosis or cellular senescence2. If these checkpoints are bypassed, continued telomere attrition can promote genomic instability and facilitate tumorigenesis7.
Malignant cells circumvent the proliferative limits imposed by telomere shortening through reactivation of telomerase, a specialized complex that elongates telomeric DNA during genome duplication8. This enzyme functions as a ribonucleoprotein complex with reverse transcriptase activity, playing a central role in preserving chromosome-end integrity and maintaining genomic stability9. In human cells, the holoenzyme comprises two principal components: a catalytic protein subunit with reverse transcriptase activity (TERT) and an RNA molecule (telomerase RNA, TR) that provides the template sequence required for telomere repeat synthesis10. Dysregulated elevation of telomerase activity contributes to tumor initiation and progression across a broad spectrum of cancers, including those of the breast, kidney, colorectum, cervix, lung, liver, pancreas, thyroid, prostate, and bladder11,12,13. Notably, telomerase reactivation is detected in approximately 85–90% of human cancers, underscoring its pivotal role in oncogenesis14,15. As such, pharmacological inhibition of telomerase represents a compelling therapeutic strategy with broad applicability across cancer types7,16. Moreover, telomerase expression is low or undetectable in most normal somatic cells, making telomerase inhibitors potentially more selective and less cytotoxic than conventional chemotherapeutics, which often indiscriminately affect both cancerous and healthy tissues17.
BIBR1532 has been developed as a highly specific telomerase inhibitor and a potential anticancer agent18. This compound selectively targets the catalytic TERT subunit of telomerase, thereby effectively suppressing telomerase activity18. Previous research demonstrated that BIBR1532 exhibits potent antitumor efficacy across various cancer types, including ovarian cancer, chondrosarcoma, breast cancer, lung cancer, and germ cell tumors19. However, pharmacokinetic limitations, such as poor bioavailability and metabolic instability, remain significant challenges for its clinical translation20,21,22,23. Such constraints reinforce the importance of developing novel small-molecule telomerase inhibitors. In recent years, in silico approaches have emerged as powerful tools for accelerating the discovery of novel therapeutic agents, particularly during the early stages of drug development24,25. Compared with conventional experimental screening methods, which are typically time-consuming and resource-intensive, computational strategies enable the rapid screening of large compound libraries. Moreover, techniques such as molecular docking, molecular dynamics simulations, and binding free energy calculations provide mechanistic insights into protein–ligand interactions at the atomic level26,27. These approaches support the identification of potential lead compounds and allow the evaluation of binding stability, conformational dynamics, and interaction mechanisms prior to experimental validation. Importantly, integrating molecular docking with molecular dynamics simulations improves prediction reliability by accounting for protein flexibility and solvent effects, which are often neglected in static docking models28,29. In addition, in silico methods help reduce failure rates in drug discovery by prioritizing compounds with favorable pharmacokinetic and toxicity profiles through early-stage ADMET prediction30,31. Therefore, such strategies represent a cost-effective and efficient alternative to traditional high-throughput screening methods, particularly for identifying natural products as potential inhibitors of key therapeutic targets such as telomerase.
Over the past decade, increasing attention has been directed toward herbal species and natural extracts as alternative or complementary approaches for cancer treatment. Numerous natural compounds have been identified with the capacity to inhibit telomerase activity32,33,34. Owing to their structural diversity and pharmacological richness, medicinal herbs represent a promising reservoir for the discovery of novel telomerase-targeting agents. In Vietnam, Celastrus hindsii is a widely used medicinal plant in traditional medicine, particularly in northern provinces such as Quang Ninh and Ninh Binh35. In traditional practices of the Muong ethnic group, C. hindsii is employed to treat a variety of conditions, including chronic diseases, inflammation, ulcers, and cancer36,37. However, its use remains largely empirical, and the bioactive compounds responsible for its purported anticancer effects, as well as their molecular mechanisms, have not been scientifically characterized. To date, no studies have investigated the telomerase-inhibitory potential of C. hindsii. In this study, we performed virtual screening of natural compounds derived from C. hindsii to identify candidates targeting telomerase. This computational approach facilitates the rapid identification of compounds with strong and stable binding affinities to telomerase, thereby highlighting promising candidates for further development as telomerase inhibitors.
MATERIALS AND METHODS
Preparation of protein and compounds
Natural compounds present in C. hindsii were identified through the LOTUS database38. Following compound retrieval, a rigorous screening process was performed to identify potential candidates. As a result, thirteen compounds satisfying the criteria of Lipinski’s Rule of Five were shortlisted for virtual screening analysis. Their three-dimensional conformations were constructed in Chem3D Pro 12.0 (CambridgeSoft, USA) employing the MM2 force field. Subsequently, energy minimization was performed using OpenBabel39 by applying the MMFF94 force field.
In this study, the structure of telomerase in complex with its high-affinity inhibitor BIBR1532 was employed as the reference model18. This model was selected due to its well-resolved crystal structure and the presence of a co-crystallized inhibitor, which provides a clearly defined binding site, making it particularly suitable for in silico investigations. Such a model facilitates the identification of key amino acid residues involved in ligand binding and improves the accuracy of docking simulations, thereby enabling the construction of a reliable simulation system40,41. The crystal structure of the telomerase + BIBR1532 complex (PDB:5CQG)18 was obtained from the Protein Data Bank. Prior to docking simulations, the protein structure was prepared with AutoDock Tools42 following standard preprocessing procedures.
Molecular docking, predicting molecular properties, pharmacokinetics and toxicological profile
The docking pocket was defined based on the binding site of BIBR1532 within the telomerase complex. A grid box of dimensions 50 × 50 × 50 Å was defined with its center positioned at x = 25.688, y = 5.971, z = –31.580, employing a grid spacing of 0.375 Å. Docking simulations were performed using AutoDock GPU43, with 1,000 iterations per compound. Compounds exhibiting a more favorable binding free energy than BIBR1532 were shortlisted for subsequent analyses. Detailed protein–ligand contacts were subsequently analyzed using the Protein–Ligand Interaction Profiler (PLIP) server44. Molecular properties and pharmacokinetic profiles were predicted using the SwissADME platform30. Toxicological profiles were assessed using ProTox version 3.045 and the pkCSM server46.
Molecular dynamics simulation
All-atom molecular dynamics simulations were performed with GROMACS 2023.547. Ligand topologies were generated under the CHARMM framework using the SwissParam web interface48, and the complete simulation systems were constructed in accordance with the CHARMM36 force field parameter set49. Each system was explicitly solvated with the TIP3P water model and supplemented with NaCl to achieve an ionic strength of 0.15 M. Systems were first subjected to energy minimization to reach a stable energetic conformation. System equilibration proceeded sequentially in the Volume-Temperature (NVT) and isothermal–isobaric (NPT) ensembles, each for 100 ps, maintained at 300 K and 1 atm. Temperature and pressure were regulated using the V-rescale thermostat (τ = 0.1 ps) and the C-rescale barostat (τ = 2 ps), respectively. The production run was executed using an integration time step of 2 fs. All simulation settings adhered to established GROMACS guidelines for CHARMM force field. Long-range electrostatics were computed using the Particle Mesh Ewald (PME) scheme with a Coulomb cutoff of 1.2 nm, while van der Waals interactions were applied with a cutoff distance of 1.2 nm. The LINCS algorithm was employed to constrain bond lengths. Trajectories were recorded every 10 ps. Trajectory analyses were performed using built-in GROMACS utilities, encompassing evaluation of root mean square deviation (RMSD), radius of gyration (Rg), solvent accessible surface area (SASA), number of protein–ligand contacts, and the free energy landscape (FEL). Principal component analysis (PCA) was performed using the MDTraj software package50 to assess the conformational space and convergence of ligand motions within each complex.
Binding free energy and amino acid distribution calculation
Binding free energy (BFE) estimations for protein–ligand interactions were conducted using the gmx_MMPBSA software package51. The MM/GBSA scheme was employed to estimate binding energetics, as it is broadly recognized for its reliability in evaluating the thermodynamic behavior of protein–ligand complexes52,53,54. All snapshots extracted from the MD trajectories were utilized for BFE calculation to ensure statistical robustness. Binding free energies were subsequently determined according to the conventional MM/GBSA formulation:
The total free energy ΔG of each system was decomposed into four principal components:
where , and represent the van der Waals and electrostatic interaction energies, respectively. denotes the polar solvation contribution, while corresponds to the nonpolar solvation component. To further delineate the interaction mechanism, residue-level energy decomposition was conducted. This analysis facilitated the pinpointing of critical amino acid residues with substantial energetic contributions to ligand association, thereby clarifying the molecular basis underlying complex stabilization.
RESULTS
Docking results, molecular properties, and toxicity predictions
The docking validation confirmed the reliability of the re-docking protocol, as BIBR1532 achieved a RMSD of 0.71 Å relative to its crystallographic pose. The calculated free energy of binding by AutoDock GPU software for BIBR1532 was −10.02 kcal/mol (Table 1). Virtual screening of natural compounds derived from C. hindsii identified a promising candidate, designated by LOTUS accession ID: LTS0233892, which exhibited a higher predicted binding affinity compared to BIBR1532, with a free energy of binding at −12.05 kcal/mol (Table 1). The molecular representation of LTS0233892 is presented in Table S1 in the Supplementary Information.
An analysis of ligand–protein interactions revealed that BIBR1532 predominantly engages in hydrophobic interactions within the telomerase binding pocket. Specifically, seven hydrophobic interactions were identified, involving amino acid residues ARG486, PHE494, TYR551, and LEU554, alongside a salt bridge interaction with ARG486 (Table 1; Figure 1). In contrast, LTS0233892 demonstrated superior binding characteristics. This compound formed six hydrophobic interactions with residues PHE494, ILE550, TYR551, and LEU554; four hydrogen bonds with ASP493, PHE494, LYS552, and GLY553; a π-stacking interaction with PHE494; and a salt bridge interaction with ARG557 (Table 1; Figure 1). Structural representations highlighting ligand engagement within the telomerase binding pocket are provided in Figure 1.
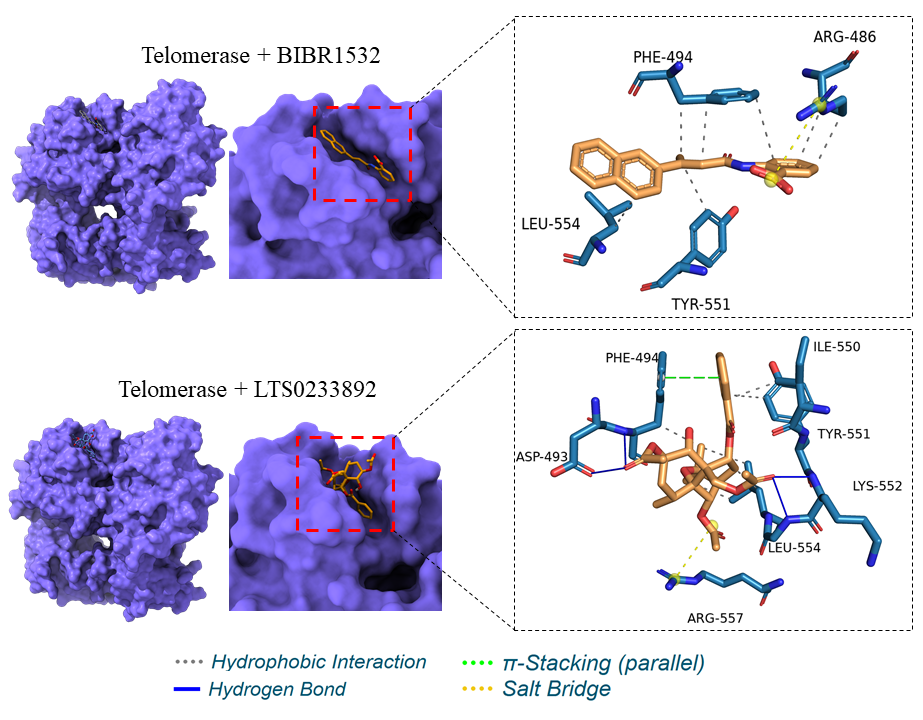
Binding interactions between BIBR1532 and LTS0233892 with the telomerase binding pocket.
Interaction analysis between telomerase and ligands (BIBR1532 and LTS0233892).
| Compounds | Free energy of binding | Hydrophobic interactions | Hydrogen bonds | π-stacking | Salt bridges |
|---|---|---|---|---|---|
| BIBR1532 | -10.02 kcal/mol |
ARG486, PHE494, TYR551, LEU554 Total interactions: 7 | - | - | ARG486 |
| LTS0233892 | -12.05 kcal/mol |
PHE494, ILE550, TYR551, LEU554 Total interactions: 6 |
ASP493, PHE494, LYS552, GLY553 Total interactions: 4 | PHE494 | ARG557 |
The analysis of molecular properties for BIBR1532 and LTS0233892 was summarized in Table 2. The findings indicated that both compounds satisfied the criteria of Lipinski’s Rule of Five and displayed favorable characteristics predictive of oral bioavailability (Table 2). Notably, LTS0233892 demonstrated superior aqueous solubility and was predicted to be unable to cross the blood–brain barrier (BBB) when compared to BIBR1532 (Table 2). Toxicity predictions were presented in Table 3. Both molecules were predicted to be non-toxic, non-irritating to the skin, and non-inhibitory to cardiac receptor systems, based on the pkCSM server results (Table 3). These findings suggested that both molecules exhibited favorable safety profiles. However, LTS0233892 exhibited superior properties, as it was not predicted to cause liver toxicity or inhibit metabolic systems (Table 3). Therefore, LTS0233892 exhibited better solubility and safety than BIBR1532 based on in silico predictions (Table 2 and Table 3).
The molecular properties and pharmacokinetic evaluation of BIBR1532 and LTS0233892.
| BIBR1532 | LTS0233892 | |
|---|---|---|
| Molecular structure | 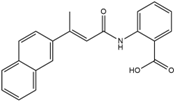 | 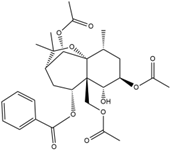 |
| Formula | C21H17NO3 | C28H36O10 |
| Molecular weight | 331.36 g/mol | 532.58 g/mol |
| Number of heavy atoms | 25 | 38 |
| Number of H-bond donor | 2 | 1 |
| Number of H-bond acceptor | 3 | 10 |
| Number of aromatic heavy atoms | 16 | 6 |
| Number of rotatable bonds | 5 | 10 |
| TPSA | 66.40 Ų | 134.66 Ų |
| Lipinski rule | Yes | Yes |
| Water Solubility | Poorly soluble | Moderately soluble |
| P-gp substrate | No | No |
| BBB permeant | Yes | No |
| GI absorption | High | High |
The toxicity profile of BIBR1532 and LTS0233892.
| Properties | BIBR1532 | LTS0233892 |
|---|---|---|
|
Hepatotoxicity (Probability) |
Active (0.61) |
Inactive (0.84) |
|
Neurotoxicity (Probability) |
Inactive (0.60) |
Inactive (0.86) |
|
Carcinogenicity (Probability) |
Inactive (0.74) |
Inactive (0.62) |
|
Mutagenicity (Probability) |
Inactive (0.53) |
Inactive (0.62) |
|
Cytotoxicity (Probability) |
Inactive (0.68) |
Inactive (0.67) |
| AMES toxicity | No | No |
| hERG inhibitor | No | No |
| Skin sensitisation | No | No |
Molecular dynamics simulations (MDS)
Telomerase mobility during MDS
Extended 200 ns molecular dynamics trajectories of telomerase in complex with BIBR1532 and LTS0233892 indicated that both systems reached stable equilibration (Figure 2). This was demonstrated by the RMSD values of the telomerase Cα backbone throughout the simulations (Figure 2A). The convergence of RMSD values after 200 ns of simulation was further illustrated by the density plot shown in Figure 2B. RMSD analysis revealed minor structural fluctuations in the telomerase+BIBR1532 complex between 25 ns and 150 ns (Figure 2A). In contrast, the telomerase+LTS0233892 complex exhibited reduced mobility and remained stable over the 200 ns simulation (Figure 2A). The average RMSD values were 0.348 ± 0.042 nm and 0.425 ± 0.068 nm for the LTS0233892 and BIBR1532 complexes, respectively (Figure 2B). These results suggest that telomerase adopted a more stable conformation upon binding to LTS0233892.
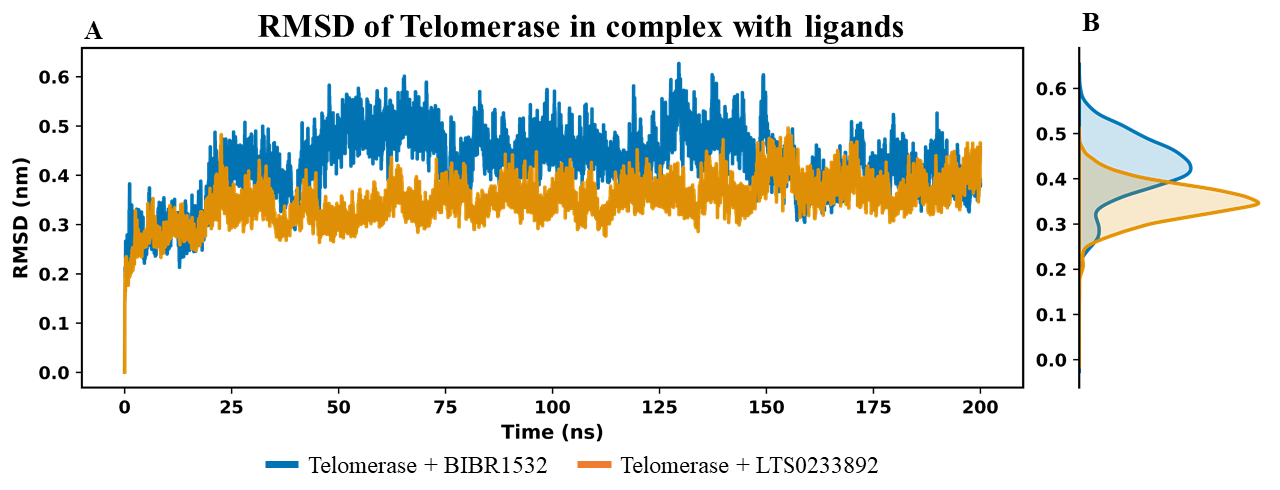
RMSD analysis of the carbon backbone of telomerase in MDS. (A) The figure shows the RMSD values of the Cɑ backbone of telomerase over the simulation time. (B) The density plot of RMSD value in 200 ns simulation.
To further investigate structural stability, the Rg value of telomerase was monitored throughout the MDS (Figure 3A). Rg is a key indicator of protein folding and compaction, with lower Rg values typically reflecting a more compact and stable conformation. Such compaction is often associated with sustained ligand binding and reduced conformational fluctuations55. Comparison of average Rg values showed that the telomerase+LTS0233892 complex maintained a more compact structure than the telomerase+BIBR1532 complex, with average Rg values of 2.932 ± 0.029 nm and 2.972 ± 0.026 nm, respectively (Figures 3B). These findings suggested that telomerase adopts a more tightly folded conformation when bound to LTS0233892. Moreover, the consistently lower Rg values observed for the LTS0233892 system indicate enhanced structural integrity and compactness throughout the simulation. In contrast, the telomerase+BIBR1532 system exhibited slightly higher Rg values, suggestive of greater conformational flexibility and reduced compaction (Figure 3A and 3B).
To complement these observations, solvent-accessible surface area analysis was performed to assess protein surface exposure (Figure 3C and 3D). Both systems exhibited stable SASA profiles with minimal fluctuations, indicating preserved global structural integrity upon ligand binding. The average SASA values were 322.685 ± 3.994 nm² for the telomerase+BIBR1532 complex and 324.629 ± 4.072 nm² for the telomerase+LTS0233892 complex (Figure 3D). Notably, the telomerase+LTS0233892 system exhibited a slightly higher SASA, indicating that the complex retains greater solvent exposure despite its increased compactness (Figure 3B and 3D). This suggests that LTS0233892 stabilizes telomerase without inducing excessive surface burial, thereby preserving a more accessible and dynamically balanced protein conformation. In conjunction with the RMSD analysis (Figure 2), these results indicate that LTS0233892 promotes a structurally stable state of telomerase, characterized by enhanced compactness alongside maintained surface accessibility. This combination highlights a favorable binding profile that supports both stability and conformational adaptability within the complex.
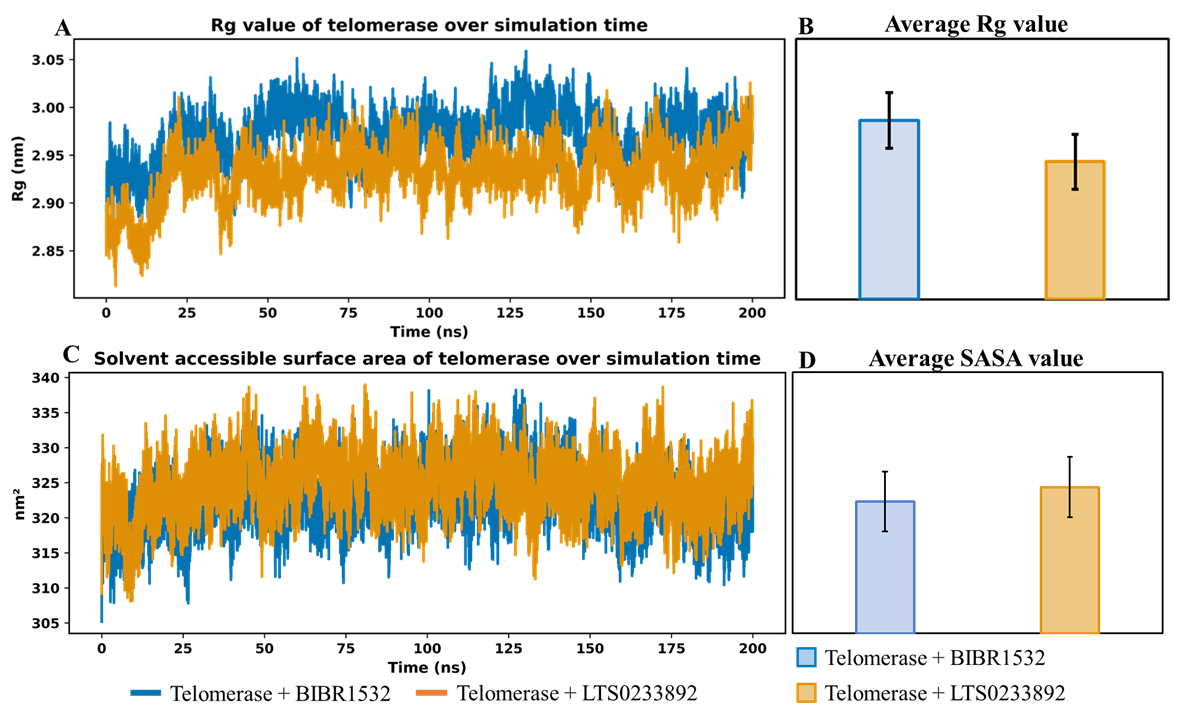
Rg values of telomerase in the MD simulation systems. (A) The line plot of Rg value of telomerase in complex with difference ligands over simulation time. (B) The average of Rg value in BIBR1532 and LTS0233892 system. (C) The line plot of SASA value of telomerase in 200 ns MDS. (D) The average of SASA value.
Ligand binding stability analysis
Ligand binding stability was assessed by evaluating the RMSD of ligand atoms relative to their initial conformation in the MDS (Figure 4). This analysis enabled characterization of the temporal stability of each ligand, whereby lower RMSD values were indicative of limited conformational deviation and more persistent interactions with the protein target56. The time-dependent RMSD profiles of each ligand are shown in Figure 4A, while the convergence of RMSD values over the 200 ns simulation is illustrated in Figure 4B. For BIBR1532, the RMSD values exhibited considerable fluctuations during the initial 30 ns of the simulation (Figure 4A), suggesting that the ligand was adjusting its binding position to establish stable interactions with telomerase. Following this equilibration phase, BIBR1532 stabilized and maintained a relatively steady conformation from approximately 50 to 200 ns (Figure 4A), achieving good convergence as shown in Figure 4B. The average RMSD value of BIBR1532 was 0.813 ± 0.181 nm. In contrast, LTS0233892 displayed greater structural stability throughout the 200 ns simulation, as reflected by consistently lower RMSD values and minimal fluctuations (Figure 4A). The average RMSD value for LTS0233892 was 0.734 ± 0.091 nm, which was notably lower than that of BIBR1532 (Figure 4B). These results indicate that LTS0233892 maintained a more stable and conformationally consistent interaction with telomerase compared to BIBR1532, suggesting a stronger and more persistent binding mode.
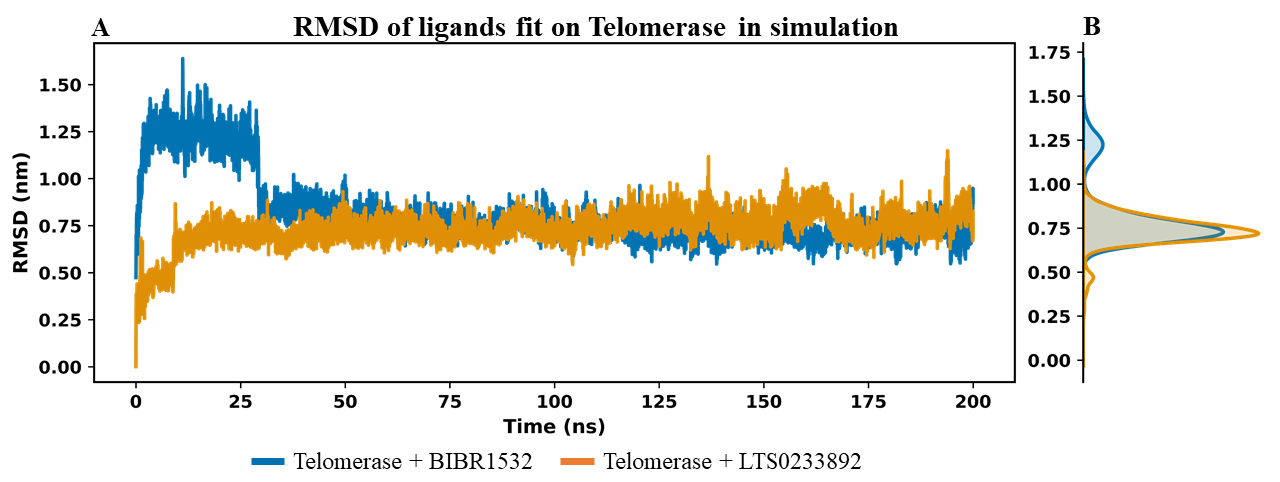
RMSD analysis of BIBR1532 and LTS0233892 molecules fit on telomerase in the MD simulation systems.(A) The RMSD value of each ligand fit on Telomerase over simulation time. (B) The density plot of ligand RMSD in 200 ns MDS.
Interaction stability was additionally evaluated by monitoring the frequency of contacts established between telomerase and the ligands over the course of the simulation. An elevated and temporally consistent contact profile is indicative of enhanced binding strength and sustained protein–ligand association. In MDS, the number of contacts between telomerase and the ligands BIBR1532 and LTS0233892 was monitored (Figure 5A). Notably, LTS0233892 formed a significantly greater number of contacts with telomerase compared to BIBR1532, with average values of 56 ± 5 and 36 ± 3, respectively (Figure 5B). This elevated contact frequency suggested that LTS0233892 established a more extensive and persistent interaction network with telomerase than the reference ligand BIBR1532.
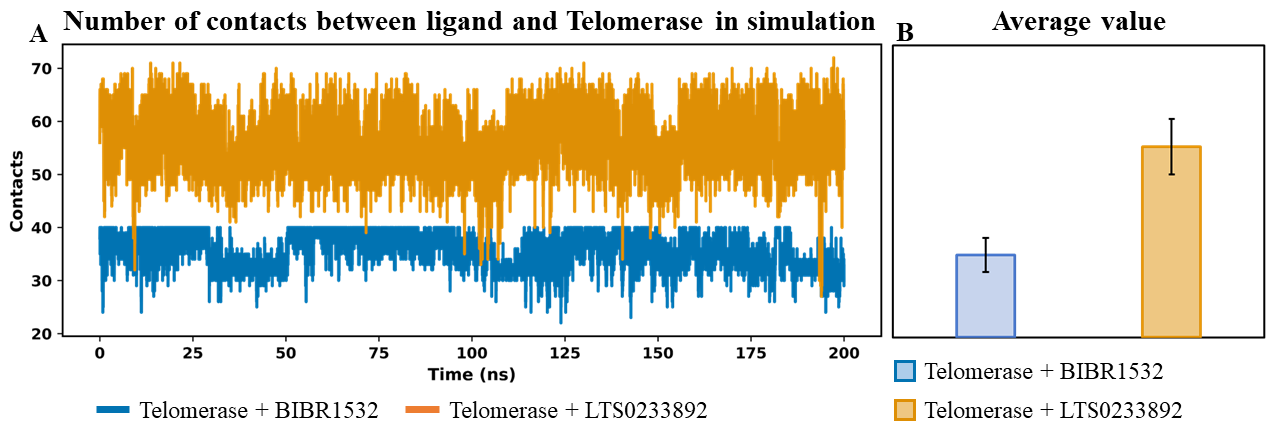
Analysis of the total number of contacts over simulation time between telomerase and ligands – BIBR1532 and LTS0233892. (A) The number of contacts between telomerase and ligands per simulation time. (B) The average value of contacts of each ligand with telomerase.
Principal component (PCA) and free energy landscape (FEL) analyses
To investigate the correlated motions and conformational dynamics of ligands during their binding to telomerase, PCA and FEL analyses were performed based on the MDS trajectories. The results for BIBR1532 are presented in Figure 6 (panels A–C), whereas those for LTS0233892 are shown in Figure 7 (panels A–C). In the PCA plots, cartesian coordinate projections represent the conformational states of each ligand at individual simulation frames, visualized as nodes, with colors indicating sampling time according to a gradient scale. The FEL is illustrated as a Gibbs free energy map. PCA was employed to characterize the dominant dynamic modes and structural fluctuations of the ligands, whereas FEL reconstruction provided insights into their thermodynamic stability throughout the simulations. In the BIBR1532 system, PCA revealed pronounced fluctuations and distinct conformational transitions during the first 100 ns of the simulation (Figure 6A). During the subsequent 100 ns, BIBR1532 exhibited reduced fluctuations and gradually converged toward a more stable conformational state after approximately 160 ns (Figure 6B). The FEL analysis of BIBR1532 further confirmed these observations, displaying two well-defined energy minima that corresponded to separate conformational states sampled during the simulation (Figure 6C). A prominent energy basin indicates a thermodynamically favorable and structurally stable ligand conformation.
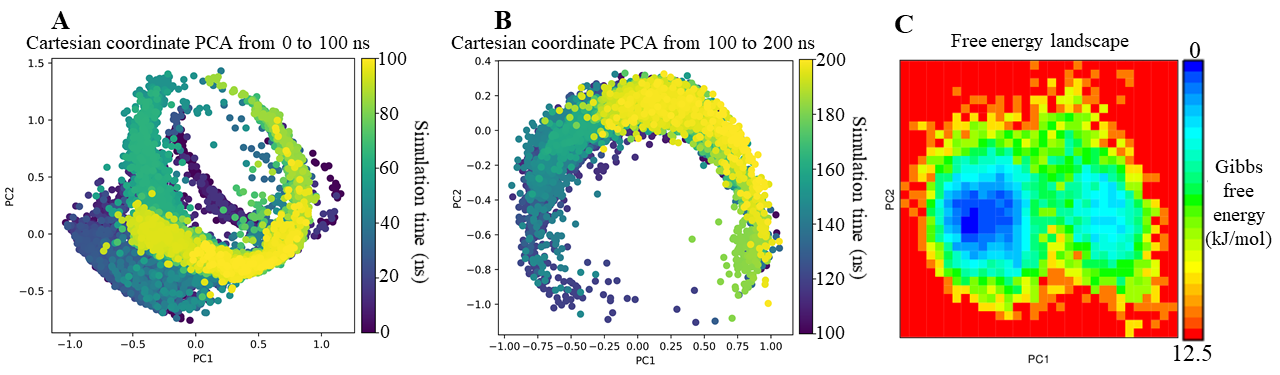
PCA and FEL analyses of BIBR1532 in complex with telomerase across MD simulation systems.(A) PCA analysis of BIBR1532 from 0 to 100 ns; (B) PCA analysis from 100 to 200 ns; (C) FEL analysis of BIBR1532.
The PCA results of the LTS0233892 system exhibited clear structural clustering, which was in stark contrast to the gradual conformational transitions observed for BIBR1532 (Figure 7). During the first 100 ns, two primary structural clusters emerged, including intermediate transition states, with convergence primarily toward a dominant cluster during the final 80 ns (Figure 7A). These clusters were maintained during the 100–200 ns interval, with transitions observed between the two states (Figure 7B). Despite these fluctuations, FEL analysis showed that LTS0233892 remained thermodynamically stable (Figure 7C). The FEL profile exhibited a single deep energy well, suggesting that LTS0233892 adopted a stable internal energy state throughout the simulations. These results indicated that the observed clustering of LTS0233892 conformers likely stemmed from minor side-chain rearrangements around a conformationally stable core, without substantially impacting its binding interaction with telomerase.
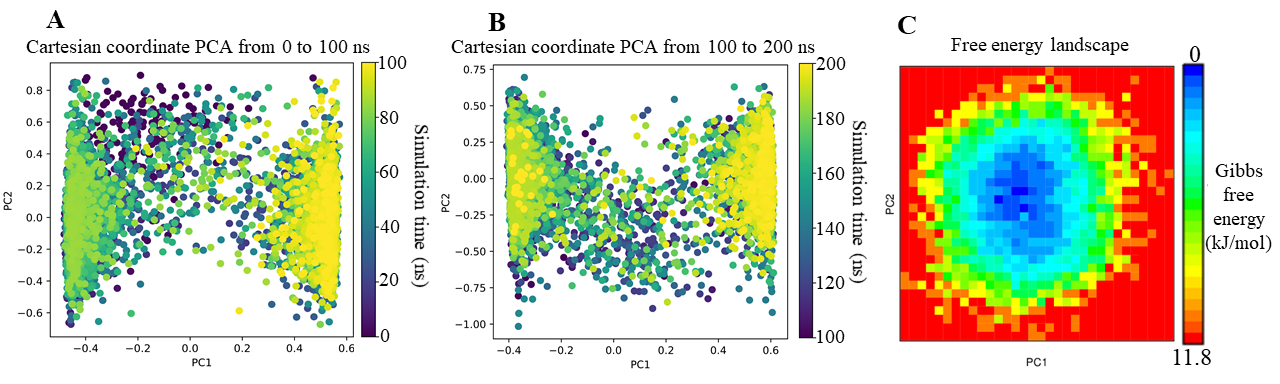
PCA and FEL analyses of LTS0233892 in complex with telomerase across MD simulation systems. (A) PCA analysis of LTS0233892 from 0 to 100 ns simulation time; (B) PCA analysis in from 100 to 200 ns simulation time; (C) FEL analysis of LTS0233892.
Binding free energy (BFE) analysis using MM/GBSA
The interaction affinity of each ligand toward telomerase was assessed via MM/GBSA-based free energy calculations. More negative energy values correspond to stronger protein–ligand interactions. The average BFE values calculated over the 200 ns MDS revealed that LTS0233892, identified from C. hindsii exhibited stronger binding affinity within the telomerase pocket compared to BIBR1532 (Figure 8). The mean BFE values were –20.996 ± 6.262 kcal/mol for BIBR1532 and –27.390 ± 4.028 kcal/mol for LTS0233892 (Figure 8). The lower BFE of LTS0233892 indicated a stronger binding tendency toward telomerase compared to BIBR1532. These results suggested that LTS0233892 could serve as a candidate for further investigation as a telomerase inhibitor. Detailed calculated MM/GBSA values and their individual energy components were presented in Table S2 of the Supplementary Information.
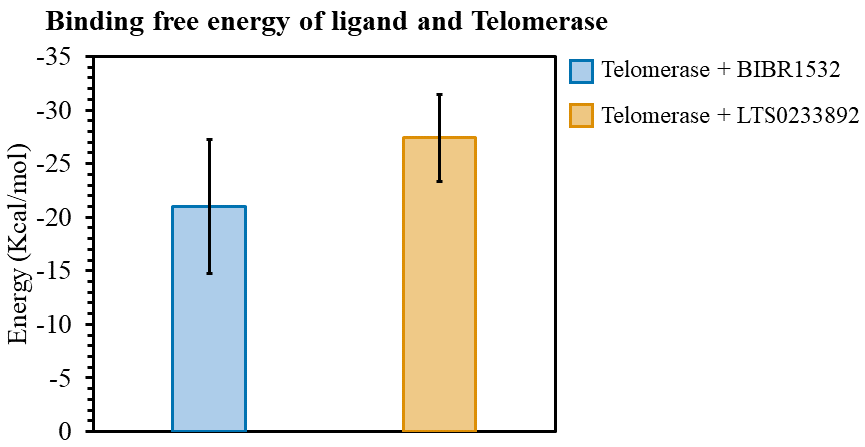
Binding free energy of BIBR1532 and LTS0233892 with telomerase during the 200 ns MDS.
To further clarify the specific interactions contributing to ligand binding, a residue-level BFE decomposition analysis was performed (Figure 9). The analysis revealed that BIBR1532 primarily interacted with two positively charged residues, ARG486 and ARG69, which acted as anchoring points within the telomerase binding pocket (Figure 9A). However, the remaining interactions were relatively weak, and the energy profile of BIBR1532 exhibited noticeable fluctuations throughout the simulation, suggesting that the ligand underwent multiple conformational adjustments before reaching a stable binding pose (Figure 9B). In contrast, LTS0233892 formed stable interactions with multiple hydrophobic residues, including PHE494, ILE497, TRP498, TYR551, and LEU554 (Figure 9C and 9D). Furthermore, the internal energy of LTS0233892 remained consistently negative during the simulations, indicating that the ligand adopted a persistent and energetically favorable binding conformation throughout the trajectory (Figure 9C). Overall, the results from BFE calculations and per-residue decomposition clearly demonstrated that LTS0233892 formed stronger and more stable interactions with telomerase than the BIBR1532 compound.
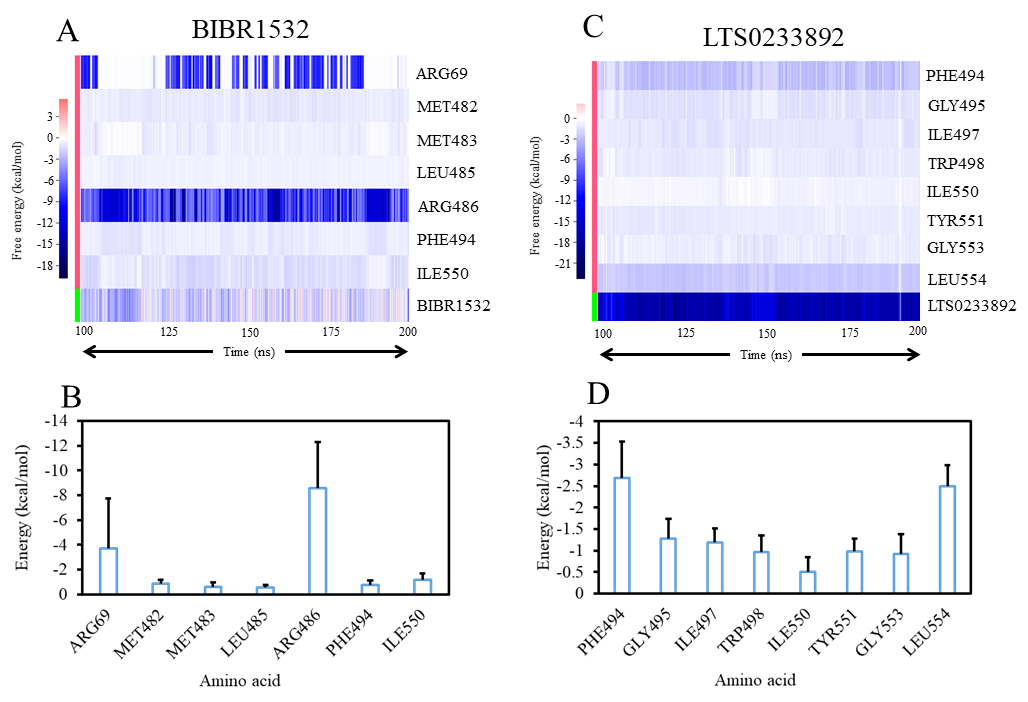
Per-residue binding free energy decomposition analysis of telomerase interactions with BIBR1532 and LTS0233892. (A) Time-dependent residue interaction energies with BIBR1532. (B) Average residue interaction energies with BIBR1532. (C) Time-dependent residue interaction energies with LTS0233892. (D) Average residue interaction energies with LTS0233892.
DISCUSSION
The expression of telomerase is recognized as a hallmark of many cancer types, making it a highly attractive therapeutic target over the past decade57,58. Telomerase has been widely recognized as an effective therapeutic target in various cancers, offering reduced side effects due to its selective targeting of cancer cells over normal cells59,60. Despite these promising findings, no telomerase-targeting drug has yet been approved by the FDA for broad-spectrum cancer treatment61. Therefore, the ongoing search for novel molecules capable of effectively inhibiting telomerase remains of critical importance. From a functional perspective, the biological consequences of telomerase inhibition have been well characterized. Compounds such as BIBR1532 have been shown to induce progressive telomere shortening, ultimately leading to growth arrest and cellular senescence in cancer cells62. Furthermore, inhibition of telomerase can disrupt telomere integrity and activate DNA damage response pathways, thereby enhancing the sensitivity of cancer cells to therapeutic interventions63. These findings underscore the central role of telomere maintenance in cancer progression and treatment response64. The compound BIBR1532 has been developed as a telomerase inhibitor by tightly binding to key amino acid residues within its binding motif. However, certain mutations within the BIBR1532 binding site of telomerase have been implicated in human diseases65,66. The residues involved in this region are known to play essential roles in ligand recognition and stabilization, highlighting their importance as anchoring sites for the rational design of more effective inhibitors.
In this context, the interaction of LTS0233892 with key residues within the active site of telomerase suggests its potential to elicit similar biological effects, likely disrupting telomerase function (Figure 9). LTS0233892 exhibits an interaction pattern comparable to that of BIBR1532, while maintaining a more distributed interaction network across the binding pocket. This observation is consistent with previous studies on telomerase inhibitors and their derivatives, which indicate that effective inhibition is governed by cooperative interactions with multiple residues rather than a single dominant contact. Such a distributed interaction pattern stabilizes the ligand within the binding pocket and reinforces the overall binding free energy landscape. In addition, this multi-residue engagement may buffer the impact of local structural perturbations, thereby reducing the likelihood of resistance arising from point mutations67,68,69. From a structural perspective, the chemical features of LTS0233892 further support its binding efficiency through its ability to form both hydrogen bonds and hydrophobic interactions (Table 1), which are known to enhance binding adaptability within a dynamic protein environment. These characteristics contribute to the sustained interaction stability observed in our molecular dynamics analyses and are consistent with established structure–activity relationship studies of telomerase inhibitors70,71. Specifically, the presence of multiple stabilizing contacts may promote persistent ligand anchoring within the binding pocket, thereby minimizing conformational fluctuations and maintaining favorable binding conformations over time. This behavior is further supported by the consistent interaction profiles and energy contributions observed throughout the simulation trajectory.
Consequently, LTS0233892 demonstrates strong potential to inhibit telomerase activity, likely through a mechanism that parallels that of BIBR1532 by directly interfering with the assembly of active enzyme subunits72. Moreover, LTS0233892 appears to outperform BIBR1532 by maintaining greater intramolecular stability and distributing its binding energy more evenly across multiple residues within the binding pocket (Figure 9). These characteristics are particularly advantageous in cancer therapy, where sustained and stable inhibition of telomerase is critical73,74. Consistent with these observations, our findings demonstrate that LTS0233892, identified from C. hindsii, has stronger binding affinity and stability than BIBR1532, which has not been previously reported. In addition, LTS0233892 exhibits predicted pharmacokinetic properties, including solubility and oral bioavailability, which may support its suitability for continued study. According to our investigation using the LOTUS database38, LTS0233892 has been reported in both C. hindsii and Salacia chinensis, a medicinal plant widely used in traditional medicine75. However, despite extensive screening of natural products, the potential interaction of LTS0233892 with telomerase has not yet been described34, and its natural abundance remains unclear. Therefore, further studies on its extraction, quantification, and structural characterization are needed. Importantly, experimental validation will be required to confirm its potential effects on telomerase activity and to assess its biological relevance.
From a broader biological perspective, beyond its predicted direct inhibitory effects, telomerase suppression may also trigger a range of downstream cellular responses. Telomerase inhibition has been associated with progressive telomere shortening, telomere dysfunction, and activation of DNA Damage Response pathways, potentially reflected by markers such as γ-H2AX, p53, and p21, which are closely linked to cell cycle arrest and cellular senescence76,77,78. Beyond its canonical role, the catalytic subunit TERT is known to participate in non-telomeric functions, including regulation of DNA repair, oxidative stress balance, and transcriptional programs. Therefore, inhibition of TERT may impair DNA repair capacity, increase reactive oxygen species (ROS) levels, and disrupt oncogenic signaling pathways79,80. However, the extent to which these mechanisms are associated with LTS0233892 remains unclear. Further experimental studies are therefore necessary to elucidate its broader biological effects, particularly the downstream consequences of telomerase inhibition and their cellular implications.
Although current computational approaches provide valuable insights, several limitations should be acknowledged. While MDS captures atomic-level motions to a certain extent, it may not fully reflect the complexity of protein–ligand interactions under physiological conditions in living systems. In addition, predicted pharmacokinetic and toxicity profiles from ADMET models are based on computational estimations and may not fully account for critical factors such as metabolism, bioavailability, and off-target effects, which remain difficult to predict accurately without experimental validation. Furthermore, the present study focuses on a single molecular target, telomerase, whereas cancer progression is governed by complex, multi-target biological networks. Consequently, potential off-target interactions and broader system-level effects of LTS0233892 have not been explored. Therefore, although our findings provide a useful preliminary framework for understanding the interaction between LTS0233892 and telomerase, they should be interpreted with caution. Further experimental studies, including biochemical assays, cell-based models, and in vivo investigations, are required to validate the predicted activity and comprehensively evaluate its therapeutic potential.
CONCLUSION
This study identifies LTS0233892, a compound derived from C. hindsii, as a novel small-molecule inhibitor targeting telomerase. Molecular dynamics–based analyses, including RMSD, PCA, FEL, and binding free energy calculations, consistently indicated a stronger binding affinity of LTS0233892 compared with BIBR1532. In parallel, pharmacokinetic and toxicity profiling suggested favorable drug-like properties and an acceptable safety profile, underscoring its potential for further development. This integrated in silico strategy provides mechanistic insights into the interactions between LTS0233892 and key residues within the telomerase binding pocket, supporting its capacity to interfere with enzymatic function. Nevertheless, these findings remain limited to computational predictions. The absence of experimental validation, together with the inherent approximations of molecular docking and molecular dynamics simulations, warrants careful interpretation. Future studies should therefore prioritize biochemical and cellular assays to validate the telomerase inhibitory activity of LTS0233892, as well as to elucidate its effects on telomere dynamics, DNA damage responses, and cancer cell proliferation. Furthermore, additional work is required to isolate and quantify this compound from C. hindsii, and to evaluate its pharmacological efficacy and safety in vitro and in vivo. These efforts will be crucial in determining whether LTS0233892 can be advanced as a viable telomerase-targeted anticancer agent.
Abbreviations
BBB: blood–brain barrier; BFE: Binding free energy; FEL: free energy landscape; MDS: Molecular dynamics simulations; NPT: isothermal–isobaric; NVT: Volume-Temperature; PCA: Principal component analysis; PDB: Protein Data Bank; PLIP: Protein–Ligand Interaction Profiler; PME: Particle Mesh Ewald; Rg: radius of gyration; RMSD: root mean square deviation; ROS: reactive oxygen species; SASA: solvent accessible surface area; TERT: reverse transcriptase; TR: telomerase RNA
ACKNOWLEDGEMENTS
This work is a part of the research project CSA.2025.063 funded by Saigon University. The authors gratefully acknowledge this financial support.
AUTHOR CONTRIBUTIONS
All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
DECLARATION OF GENERATIVE AI ANDAI-ASSISTED TECHNOLOGIES IN THE WRITINGPROCESS
The authors declare that they have not used any generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data.)
COMPETING INTERESTS
The authors declare that they have no competing interests.