
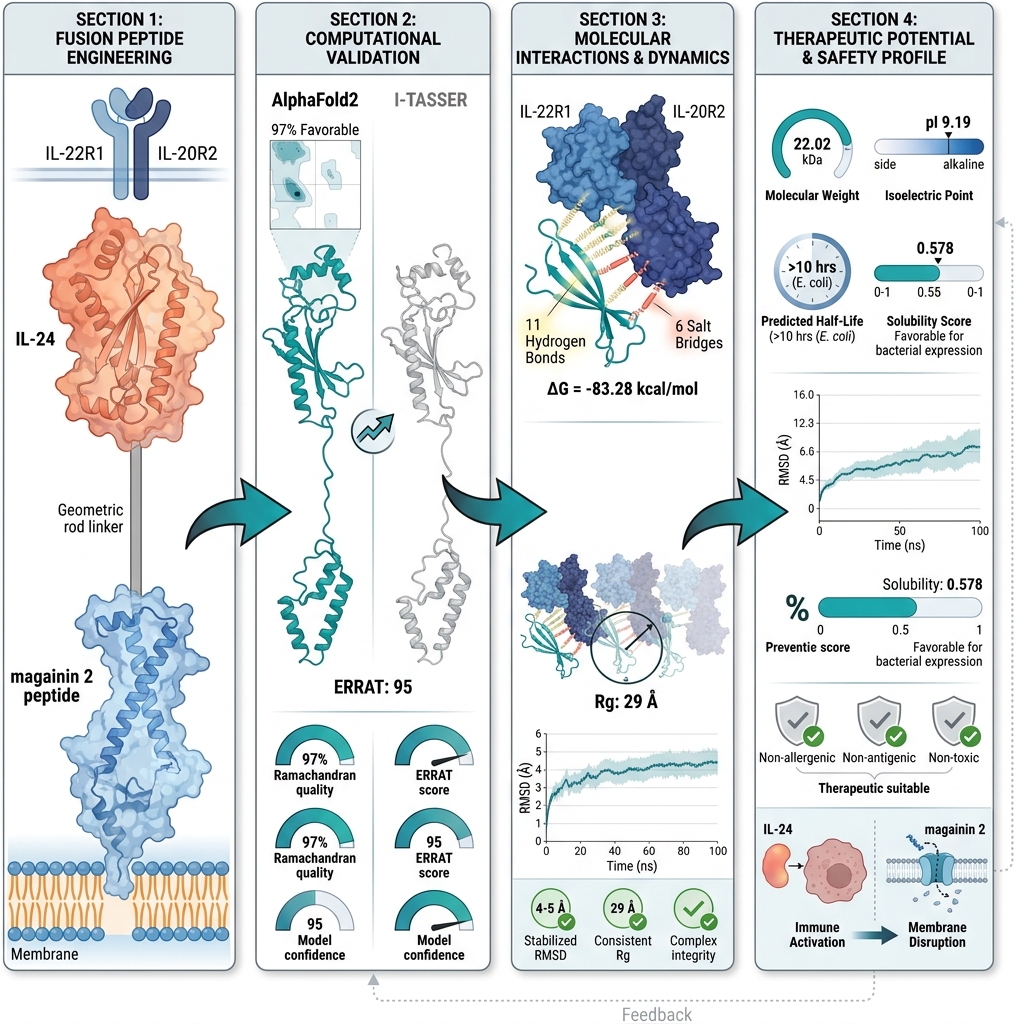

Precision-Engineered Fusion Peptide for Breast Cancer Therapy: A Molecular Docking and Simulation Study
- University Institute of Medical Lab Technology, Faculty of Allied Health Sciences, The University of Lahore -54590 Pakistan
- Department of Pharmacology, Faculty of Pharmacy and Health Sciences, University of Balochistan, Quetta, Pakistan
- Centre for Applied Molecular Biology, 87-West canal, Bank Road, University of the Punjab, Lahore-53700, Pakistan
Abstract
Introduction: Despite therapeutic advancements, breast cancer remains a formidable clinical challenge, necessitating innovative therapeutic strategies that combine high specificity with enhanced potency. To address this need, we rationally engineered a novel fusion protein that integrates the membrane-lytic activity of magainin-2 with the tumor-targeting capability of interleukin-24 (IL-24), aiming to significantly enhance selective cytotoxicity against breast cancer cells.
Methods: A 3D model of the magainin 2–IL-24 fusion protein was generated using AlphaFold2 and subsequently subjected to rigorous refinement, structural validation, and comprehensive assessment of its physicochemical properties.
Results: Structural validation via Ramachandran plot and ERRAT2 analyses confirmed the robust structural quality of the model. Molecular docking studies revealed 11 hydrogen bonds and additional intermolecular contacts, including salt bridges, between the fusion protein and its cognate receptor, indicating strong and highly specific interactions. Molecular dynamics simulations conducted over 100 ns further validated the conformational stability of the docked complex.
Conclusion: These findings highlight the fusion protein’s capacity to combine targeted delivery with membrane disruption, offering a potent dual-action mechanism. This study provides a robust scientific rationale and computational evidence that the successful in vitro expression of the magainin 2–IL-24 fusion gene could yield a promising next-generation therapeutic candidate for breast cancer treatment.
INTRODUCTION
Globally, cancer remains a leading cause of mortality, characterized by uncontrolled cell proliferation and the potential to affect virtually any organ or tissue1. In 2020, cancer accounted for approximately 10 million fatalities; among these malignancies, breast cancer exhibited a particularly alarming mortality rate. Between 2020 and 2030, the incidence of breast cancer is projected to rise markedly, with women aged 45–49 years facing the highest risk2. Current therapeutic modalities for breast cancer include surgery, radiotherapy, hormone therapy, chemotherapy, and adjuvant therapy. However, the long-term efficacy of chemotherapy is often compromised by the emergence of drug resistance mechanisms involving altered cellular processes, extracellular factors, and systemic pharmacology3. Additionally, radiation therapy generates free radicals that induce cellular dysfunction and death in both malignant and healthy tissue4,5. The persistent rise in breast cancer incidence necessitates the development of novel therapeutic agents that offer precise targeted delivery, reduced off-target toxicity, and enhanced clinical efficacy6. One promising strategy is the utilization of fusion proteins, which integrate two distinct protein domains to yield synergistic therapeutic properties. Specifically, combining an interleukin with an anticancer peptide can significantly augment the overall anti-tumor potential of the construct. Among various cytokines, interleukin-2, interleukin-15, and interleukin-24 have demonstrated potent anti-tumor activity7; however, the requirement for high-dose administration limits their clinical utility and practicality. To address this limitation, targeted drug delivery methods are increasingly adopted in oncology. Targeted delivery selectively directs therapeutics toward cancer cells, mitigating the development of drug resistance while enhancing overall pharmacological efficacy8. Natural peptides serve as advantageous anti-tumor agents due to their enhanced specificity and reduced toxicity. The fusion of homing peptides with lytic peptides in cancer pharmacotherapy offers substantial benefits, including high selectivity, targeted delivery, improved solubility, and attenuated systemic toxicity9. Consequently, the therapeutic effectiveness of such recombinant proteins is significantly enhanced, with synergistic impacts reportedly increasing by 10-to-20 times10.
As a member of the interleukin-10 family of cytokines, interleukin-24 (IL-24) is involved in immune system regulation by signaling molecules, inducing inflammation, and modulating cellular function through the alteration of gene expression11. Secreted primarily by T-cells and monocytes, IL-24 can suppress tumor growth by activating the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway. Furthermore, it can halt protein translation by inducing endoplasmic reticulum (ER) stress mechanisms within breast cancer cells9.
The African clawed frog (Xenopus laevis) secretes a lytic peptide from its skin called magainin 2, comprising 23 amino acids with a molecular mass of 2466.9 g/mol12. Upon contact with the cancer cell membrane, magainin 2 and its analogs operate via a "carpet-like" mechanism, attaching to the phospholipid head groups. This interaction induces severe disruption of the lipid bilayer architecture, resulting in robust anti-tumor activity13. Magainin 2 offers distinct mechanistic advantages, including potent membrane-lytic activity coupled with low hemolytic toxicity and the selective disruption of cancer cell membranes, distinguishing it from other lytic peptides14. This study aims to fuse the magainin 2 peptide with IL-24 via a rigid linker to harness the synergistic effects of this chimeric peptide against breast cancer cell receptors (IL-24 receptors are frequently overexpressed on these cells), thereby enhancing its therapeutic potential. The incorporation of a rigid linker is intended to maintain structural separation between the two functional domains, minimize steric interference, preserve independent folding, and enhance the overall stability and functional efficiency of the fusion construct.
The magainin 2–IL-24 fusion peptide was evaluated for structural quality and validation, docked to the IL22R1–IL20R2 heterodimeric receptor on breast cancer cells, and subsequently analyzed for interactions, dynamic stability through simulation, and in silico expression potential. Computational approaches, such as molecular docking and molecular dynamics simulations, have become essential preliminary tools in therapeutic protein design, allowing for the detailed evaluation of structural stability, receptor binding, and dynamic behavior prior to in vitro or in vivo experimental studies15,16. These in silico methodologies provide a solid foundation for drug design by identifying the most promising candidates efficiently.
MATERIALS AND METHODS
Chimeric protein construction
The FASTA sequence for IL-24 was retrieved from the Universal Protein Knowledgebase (Accession number: Q13007), while the sequence for the lytic peptide magainin 2 was obtained from the previously reported work of Hoskin17. The 3D structure of the heterodimeric IL-24 receptor was acquired from the Protein Data Bank (PDB ID: 6DF3). To isolate the mature fragment of the IL-24 peptide, amino acids 1-56 from the amino terminus were removed. The remaining peptide was fused to magainin 2 via a previously reported rigid linker sequence (EAAAKEAAAKEAAAK)18 to engineer the final fusion construct.
Prediction of protein secondary structure
The secondary structure of the magainin 2-IL-24 fusion protein was predicted using the GOR-IV server. This method analyzes the FASTA sequence of the chimeric protein to provide insights into its secondary structural elements, including alpha-helices, random coils, beta-sheets, coiled coils, low-complexity regions, extended strands, and structurally disordered regions19,20.
3D structure modeling and refinement of magainin 2-IL-24 fusion protein
Three-dimensional (3D) structural modeling was performed using both the AlphaFold2 and I-TASSER (V5.1) online servers. Employing two distinct methodologies enhanced the accuracy and reliability of the predicted structure and facilitated the selection of the most optimal model for subsequent analyses. I-TASSER (V5.1) primarily utilizes a template-based modeling approach, initiating the process by identifying structurally similar proteins with known 3D structures from the Protein Data Bank (PDB) using threading and sequence alignment techniques21,22. AlphaFold2, developed by DeepMind, relies on deep learning techniques, employing a neural network architecture designed to predict inter-residue distances within the protein sequence. Both servers require only the FASTA sequence as an input to construct a 3D model23. The generated models were further refined to minimize energy, optimize side-chain conformations, and improve stereochemistry and bond angles using the Galaxy Refine (V2.0) online server24.
Structural validation of the fusion protein
The quality of the constructed models was evaluated using multiple computational tools. For an overall accuracy assessment, the ProSa-web tool was employed to calculate the Z-score, which provides the global quality of the 3D model and estimates its structural alignment with experimentally resolved proteins25. Next, the overall quality and reliability of the model were assessed through the ERRAT2 graph26. Lastly, the 3D model was validated for its stereochemical integrity by generating a Ramachandran plot.
Prediction of toxic effects, immunogenicity, and allergen potential
The ToxinPred server was utilized to predict regions within the submitted sequence containing toxic amino acid residues27. Allergenicity was assessed using AllerTOP, which operates based on the similarity index of protein regions with known epitopes28. The VaxiJen server was employed to compute antigenicity based on physicochemical properties29, utilizing a 0.5 threshold to differentiate between antigenic and non-antigenic properties. These predictive analyses served as a critical foundation for evaluating the safety profile of the fusion protein as a potential drug candidate.
Docking analysis and interaction studies
Blind molecular docking was performed using the ClusPro 2.0 online server16,30. The 3D structure of the heterodimeric receptor was downloaded and prepared using the PyMOL molecular graphics system, which involved the removal of endogenous IL-24, ligands, and water molecules to ensure a purified receptor structure. The PDB-formatted structures of the fusion protein and its receptor were uploaded to the ClusPro 2.0 server, and docking was performed using default parameters. The resulting docked complex was subjected to an in-depth analysis of protein-protein interactions using PDBsum and PDBePISA, enabling the identification of critical features such as binding affinity, interacting interfaces, hydrogen bonds, solvation energy (kcal/mol), and salt bridges within the docked complex31.
Molecular dynamics simulation
The top-ranked docked complex was subjected to molecular dynamics (MD) simulation using NAMD v2.14, with trajectory visualization performed in VMD v1.9.332. System preparation and topology generation were carried out using AmberTools21, employing the ff14SB force field for the protein complex33. Missing hydrogen atoms were added, and protonation states were assigned to be consistent with physiological pH. The complex was solvated in an explicit TIP3P water model within an orthorhombic periodic box extending 10 Å beyond the protein in all directions. Counter ions (Na⁺ and Cl⁻) were added to neutralize the system and achieve physiological ionic strength.
Prior to the production simulation, the system underwent energy minimization for 10,000 steps using the conjugate gradient algorithm to resolve steric clashes. This was followed by a two-stage equilibration process: an NVT ensemble equilibration at 310 K using Langevin dynamics for temperature control, and an NPT ensemble equilibration for 1 ns at 1 atm pressure, maintained using the Nosé–Hoover Langevin piston method. The production MD simulation was then performed for 100 ns under periodic boundary conditions. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method, and covalent bonds involving hydrogen atoms were constrained using the SHAKE algorithm. Trajectory analyses were conducted using the bio3d package (v2.4-0) in R (v4.x)34, where structural stability and conformational dynamics were evaluated through root-mean-square deviation (RMSD), radius of gyration (Rg), and root-mean-square fluctuation (RMSF). Stability was assessed based on RMSD convergence and plateau behavior during the latter half of the simulation.
Expression potential assessment in E. coli
To predict the soluble expression of the fusion protein in E. coli, the SoluProt online server was utilized. This computational tool was selected based on its excellent accuracy compared to other existing tools, as demonstrated by Ghomi et al.35. By leveraging SoluProt, we were able to generate informed predictions expected to significantly enhance the efficiency and success of future in vitro protein expression experiments, ensuring optimal conditions for high-yield expression.
RESULTS
Engineering of magainin 2-IL 24 fusion protein
The FASTA sequences encoding IL-24 and magainin 2 were retrieved from the National Center for Biotechnology Information (NCBI) database and from a previously published study, respectively. To engineer a single polypeptide chain comprising 193 amino acids, these sequences were fused via a rigid linker incorporating the amino acid sequence EAAAKEAAAKEAAAK (Figure 1A). This rigid linker plays a crucial role in preventing aberrant disulfide bond formation between the distinct functional domains of the fusion protein. To facilitate downstream molecular cloning for in vitro expression studies, specific restriction enzyme cleavage sites were incorporated at both the N- and C-termini.
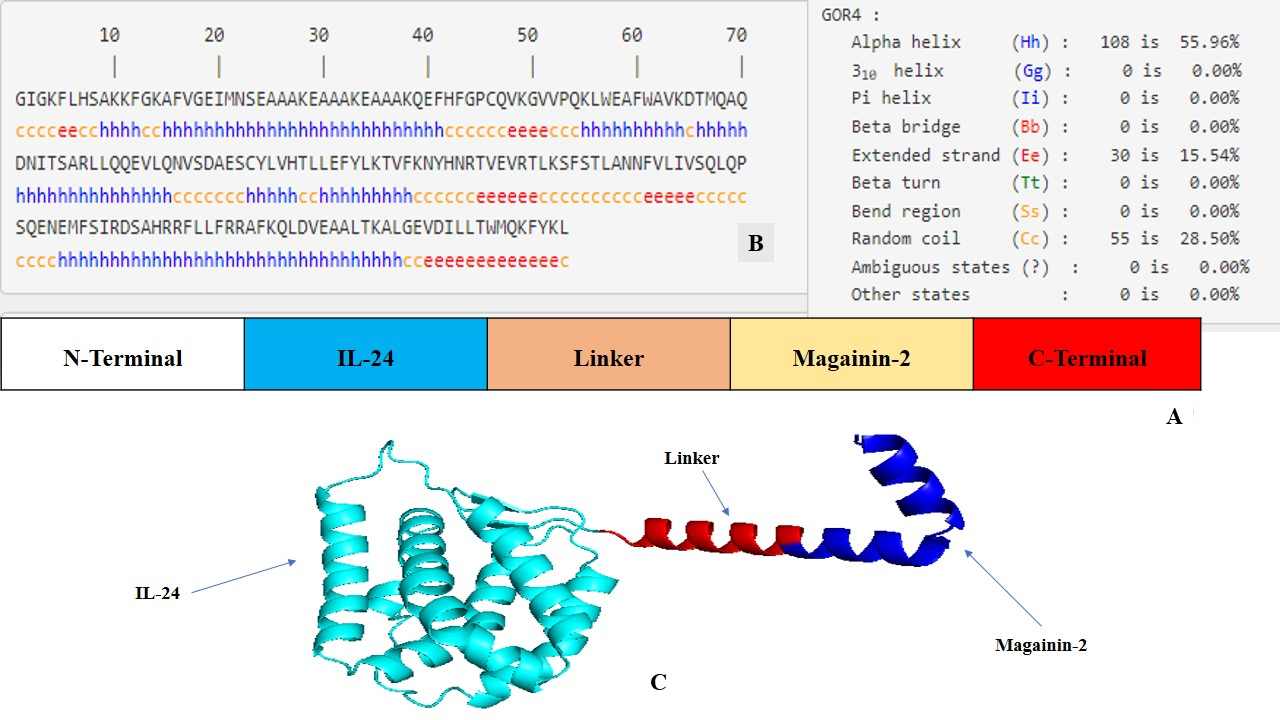
Structural design and computational modeling of the magainin 2–IL-24 fusion protein.(A) Schematic representation of the engineered chimeric construct, illustrating the linear arrangement of the IL-24 domain, the rigid linker, and the magainin 2 peptide. (B) Secondary structure prediction of the fusion sequence generated using the GOR IV server, detailing the distribution of alpha-helices, extended strands, and random coils. (C) The optimal three-dimensional (3D) structure of the fusion protein predicted by AlphaFold2, demonstrating the spatial separation and independent folding of the functional domains. The model was visualized using PyMOL (v 3.1).
Secondary structure evaluation of chimeric proteins
Secondary structure prediction of the magainin 2-IL-24 chimeric protein, conducted using the GOR IV server, provided valuable insights into its structural composition and potential functional roles. Notably, the analysis revealed that a substantial portion of the protein (55.96%) adopts an alpha-helical conformation. This abundance of alpha helices signifies a high degree of structural stability, which is pivotal for the protein's overall functionality and therapeutic efficacy. Moreover, the presence of extended strands and beta-sheets (15.54%) adds structural diversity that may facilitate specific functional interactions and enhance adaptability to various molecular environments. Additionally, the remaining 28.50% random coil conformation provides critical flexibility, which is essential for the construct's dynamic biological functions (Figure 1B).
3D structure modeling of magainin 2-IL 24 fusion protein
To generate accurate three-dimensional representations, the chimeric protein's FASTA sequence was initially submitted to both the AlphaFold2 and I-TASSER (V5.1) online servers. AlphaFold2, recognized for its exceptional side-chain modeling accuracy, generated five models ranked by local model quality metrics; the top-ranked model was selected for subsequent analysis. I-TASSER (V5.1), employing its C-score as a confidence measure, similarly produced five candidate models. The model exhibiting the highest C-score (0.97), corresponding to a TM-score of 0.796 and an RMSD of 0.34, was chosen for comparison. The top models from both computational platforms were then compared to determine the most precise and stable configuration of the chimeric protein. A comprehensive evaluation based on structural quality, validation metrics, and Ramachandran plot scores demonstrated that the AlphaFold2-generated model was significantly superior to the I-TASSER (V5.1) model across overall scoring parameters. Consequently, the AlphaFold2 structure was selected for all downstream analyses (Figure 1C).
Reliability and validation-based selection of a chimeric protein model
The comparison of homology models generated by AlphaFold2 and I-TASSER (V5.1) involved the evaluation of several critical parameters to assess structural quality and reliability. The Ramachandran plot, a crucial tool for assessing stereochemical quality, revealed that both models possessed a high proportion of amino acids in the favorable region (97% for AlphaFold2 and 92% for I-TASSER), indicating energetically favorable phi and psi conformations. However, AlphaFold2 exhibited superior stereochemistry (Figure 2A). ERRAT2 analysis, which evaluates non-bonded atomic interactions, yielded an ERRAT score of 95 for the AlphaFold2 model, compared to a slightly lower score of 94% for the I-TASSER (V 5.1) model, suggesting better agreement with high-resolution structural data (Figure 2B). The Z-score, which evaluates overall model quality relative to experimentally determined structures, was negative for both models, reflecting favorable native-like folding. The AlphaFold2 model yielded a Z-score of -5.84 compared to the I-TASSER (V 5.1) model score of -6.02 (Figure 2C). Collectively, based on Ramachandran stereochemistry, ERRAT scores, and global Z-scores, the AlphaFold2 model proved to be more reliable and accurate, justifying its selection for further analysis and applications involving the fusion protein structure.
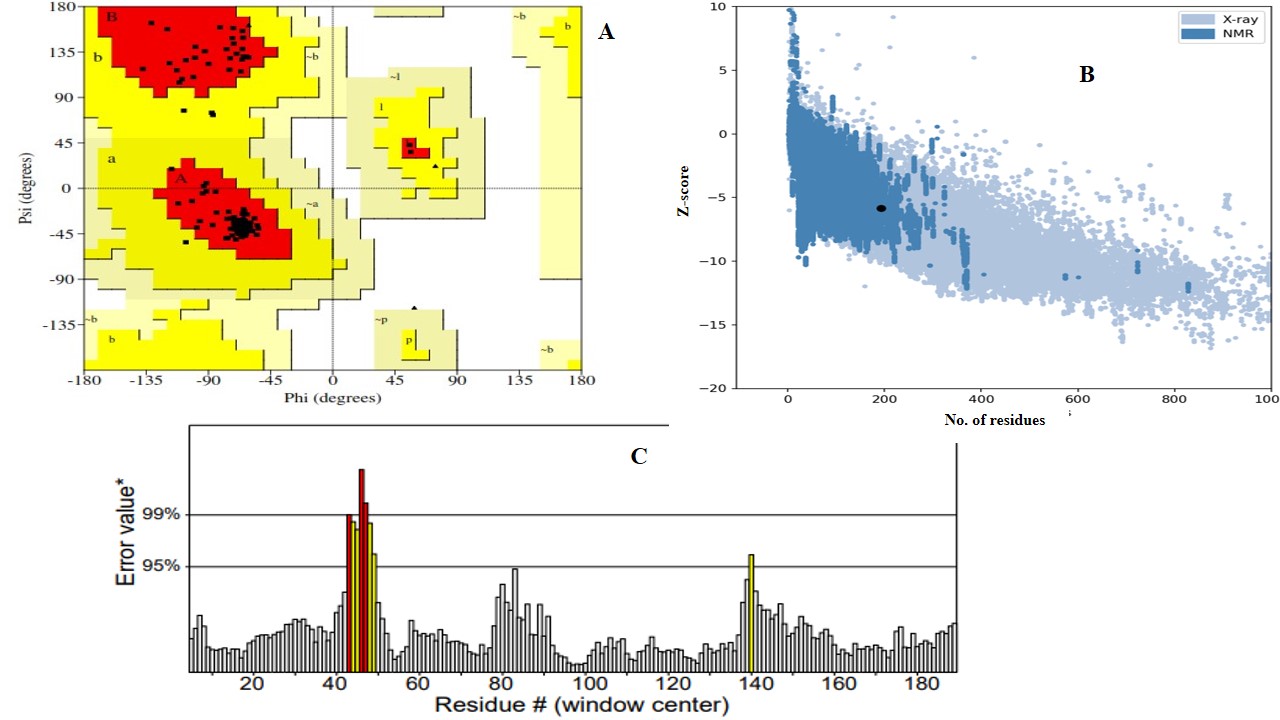
Structural validation and quality assessment of the AlphaFold2-generated magainin 2–IL-24 fusion protein model.(A) Ramachandran plot evaluating the stereochemical quality of the 3D model, demonstrating that 97% of the amino acid residues are located in energetically favorable regions. (B) ProSA-web plot indicating a global model quality Z-score of -5.84, reflecting a favorable, native-like fold relative to experimentally resolved protein structures. (C) ERRAT2 evaluation plot analyzing non-bonded atomic interactions, yielding a high overall quality factor of 95 to confirm structural reliability.
Physicochemical characteristics and solubility prediction
The physicochemical properties of the chimeric fusion protein provide valuable insights into its potential as a therapeutic agent. These properties were estimated using the ProtParam server (Table 1). The fusion protein consists of 193 amino acids with a molecular weight of 22.02 kDa, indicating its substantial size and complexity. The isoelectric point (pI) of 9.19 suggests that the protein is relatively basic. Additionally, a high extinction coefficient of 22,585 M⁻¹ cm⁻¹ indicates that it can be effectively quantified via spectrophotometric assays, facilitating further analysis and development. Notably, the protein contains 20 negatively charged residues and 25 positively charged residues, which may influence electrostatic interactions during physiological processes. Moreover, the estimated half-life of over 10 hours in E. coli, along with an instability index of 36.78, suggests that the protein is thermodynamically stable and persistent within a biological system. A high aliphatic index (88.50) and a negative Grand Average of Hydropathicity (GRAVY) score (-0.147) indicate an optimal balance of hydrophilicity and hydrophobicity, facilitating interaction with cellular components. On the Protein-sol online server, the selected model exhibited a scaled solubility value of 0.478, which exceeds the threshold of 0.45, indicating that the fusion protein is likely soluble—a critical factor in drug formulation and delivery. Adequate solubility ensures the protein is readily available for interactions with target cancer cells and can be administered through various pharmaceutical routes. These properties collectively emphasize the structural viability of the magainin 2-IL-24 fusion protein as a stable, soluble candidate for anti-cancer therapeutics, highlighting its potential for targeted interactions and stability within biological systems, making it a promising candidate for further investigation and therapeutic development.
Physiochemical properties of fusion construct.
| Properties | Values |
|---|---|
| Number of amino acids | 193 |
| Theoretical pI | 9.19 |
| Molecular weight | 22023.45 |
| Instability index | 36.78 |
| Aliphatic index | 88.5 |
| Total positively charged amino acids | (Arg + Lys): 25 |
| Total negatively charged amino acids | (Asp + Glu): 20 |
| Grand average of hydropathicity (GRAVY) | -0.147 |
| Predicted half-life | >10 hours ( |
| Coefficient of extinction (in M-1 cm-1 at 280 nm) | 22585 |
Evaluation of allergenicity, antigenicity, and toxicity
The comprehensive assessment of allergenicity, antigenicity, and toxicity is an essential step in evaluating the clinical safety of therapeutic proteins. In this study, AllerTOP predicted the fusion construct to be non-allergenic, indicating a low probability of inducing hypersensitivity reactions upon systemic administration. VaxiJen analysis, utilizing a threshold of 0.5, classified the protein as non-antigenic, suggesting it is unlikely to provoke adverse immune responses—a characteristic that is highly desirable for therapeutic applications. Furthermore, ToxinPred evaluations confirmed its non-toxic nature, substantively reinforcing the overall safety profile of the chimeric protein. Collectively, these in silico predictions support a highly favorable safety profile, providing a robust rationale for future experimental and in vivo studies to validate the therapeutic applicability of this chimeric construct.
Expression prediction in E.coli
Computational analysis utilizing SoluProt 1.0 predicted a solubility score of 0.578. By exceeding the predefined threshold value of 0.5, this result strongly suggests a favorable probability of soluble protein expression within an E. coli host system. While this in silico prediction indicates high feasibility for successful expression, it does not definitively confirm soluble production yields under in vitro experimental conditions. Consequently, rigorous empirical validation remains imperative to confirm the actual expression levels and solubility profile of the construct within a bacterial system.
Docking analysis
The docking study between the magainin 2-IL-24 fusion protein and its heterodimeric receptor provided critical insights into their binding affinity and structural interaction. By employing ClusPro 2.0 for protein-protein docking, the mechanism of interaction between the fusion protein and its receptor was elucidated, yielding a rigorous assessment of their binding affinity. The selection of the top docked structure was predicated on scoring criteria, including electrostatic energies and van der Waals attractions (Figure 3). Notably, the major interactions observed during protein-protein docking were driven by electrostatic and van der Waals forces, which yielded promising and reliable results36. The robust docking methodology inherent to ClusPro 2.0 ensures high accuracy in the binding affinity evaluation. The generation of ten distinct dock models, each with unique weighted scores, allowed for a comprehensive assessment of binding energetics. The optimized balanced model exhibited a center-weighted score of -868.9 and a minimum energy value of -898.2 kcal/mol, reflecting a highly stable and strong binding interaction (Table 2). These results signify the robust potential of the magainin 2-IL-24 fusion protein to interact effectively with the IL-24 heterodimeric receptor, which is a pivotal step in its development as a candidate for anti-cancer therapeutics. In this analysis, the center score represents the average energy of surrounding structures, while the weighted lowest-energy score reflects the most stable conformation within that specific cluster.
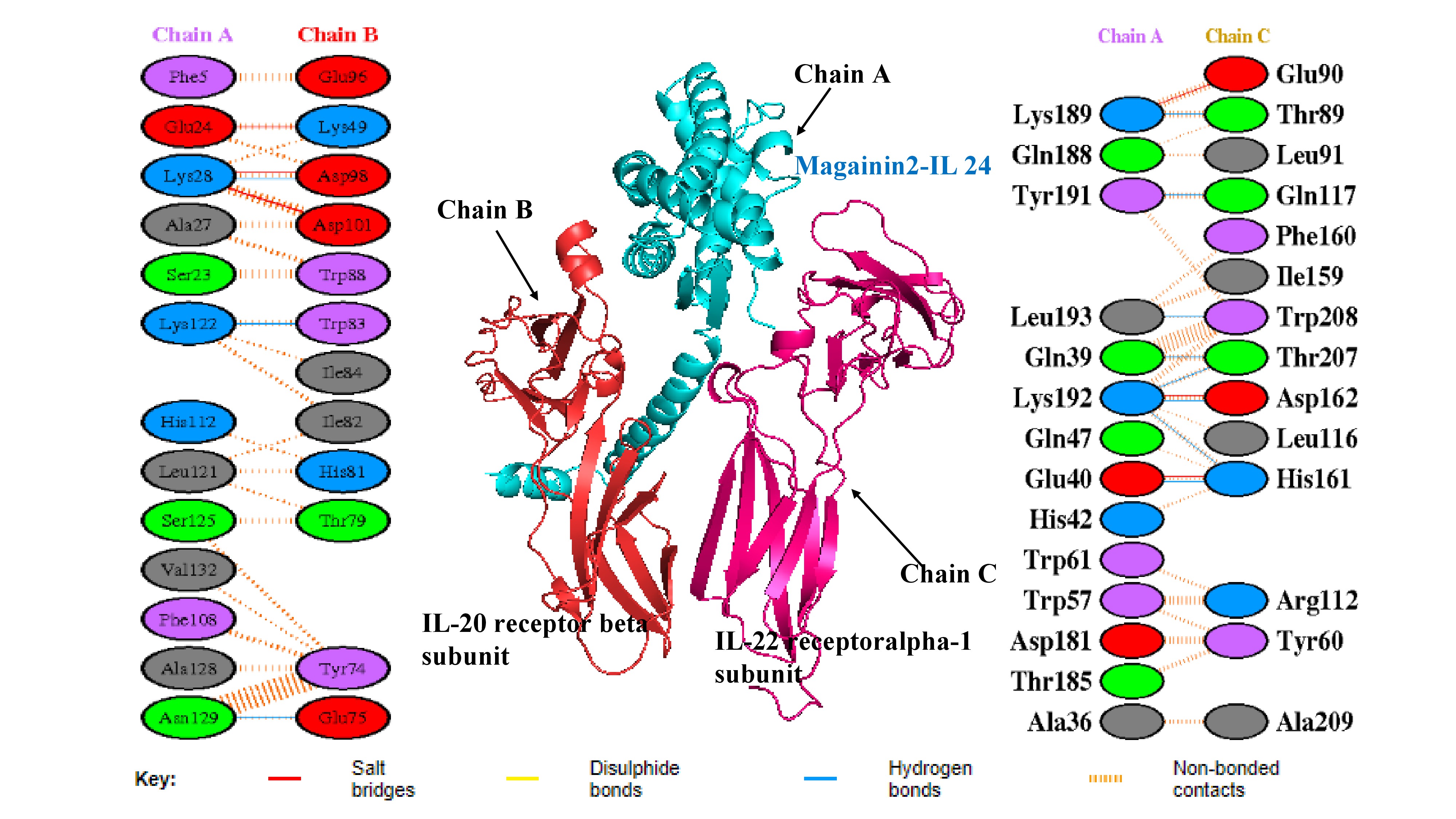
Molecular docking complex and interfacial interaction networks between the magainin 2–IL-24 fusion protein and the heterodimeric receptor. The central 3D structural model illustrates the optimal docking pose, depicting the fusion protein (Chain A, cyan) bound to the IL-20R2 (IL-20 receptor beta, Chain B, red) and IL-22R1 (IL-22 receptor alpha-1, Chain C, magenta) subunits. Flanking the central structure are PDBsum-generated 2D interaction maps detailing the specific residue-level contacts at the binding interfaces. The left panel maps interactions between the fusion protein and the IL-20R2 subunit, while the right panel highlights interactions with the IL-22R1 subunit. Key stabilizing forces are denoted by colored lines, including salt bridges (red), hydrogen bonds (blue), and non-bonded contacts (orange dashes).
Balanced coefficient scores of top dock complexes.
| Cluster | Members | Characteristic | Weighing Score |
|---|---|---|---|
| 00 | 81 | Center | -868.8 |
| Low Energy | -898.2 | ||
| 01 | 81 | Center | -859.9 |
| Low Energy | -863.8 | ||
| 02 | 46 | Center | -789.2 |
| Low Energy | -849.1 | ||
| 03 | 45 | Center | -728.2 |
| Low Energy | -907.5 | ||
| 04 | 36 | Center | -867.6 |
| Low Energy | -867.4 | ||
| 05 | 35 | Center | -825.4 |
| Low Energy | -910.8 | ||
| 06 | 32 | Center | -723.0 |
| Low Energy | -927.5 | ||
| 07 | 29 | Center | -727.7 |
| Low Energy | -847.1 | ||
| 08 | 25 | Center | -719.0 |
| Low Energy | -816.2 | ||
| 09 | 24 | Center | -800.6 |
| Low Energy | -800.6 | ||
| 10 | 22 | Center | -776.2 |
| Lowest Energy | -861.4 |
Residue interactions analysis
In recent years, advancements in molecular modeling software and computational tools have greatly accelerated drug discovery efforts, particularly in elucidating the complex protein-protein interactions (PPIs) that are relevant to various diseases, including cancer. Historically, PPIs were often considered "undruggable" due to their challenging binding interfaces; thus, the investigation of such interactions represents a critical frontier in modern pharmacological research37. Computational analysis of the docked complex, facilitated by the PDBePISA and PDBsum servers, provided valuable insights into the binding interface, which was characterized by the presence of 17 intermolecular interactions, encompassing 6 salt bridges and 11 hydrogen bonds. Specifically, regarding the fusion protein's interaction with the IL-22R1 subunit, 11 distinct bonds were predicted, comprising 3 salt bridges and 8 hydrogen bonds. This interaction is particularly robust, with interfacial residues spanning an area of 695.2 Ų and contributing to a stabilization energy of -6.5 kcal/mol. Conversely, the interaction between the fusion protein and the IL-20R2 subunit resulted in 6 bonds, consisting of 3 salt bridges and 3 hydrogen bonds. This interaction spans an interface of 679.8 Ų and is associated with a stabilization energy of -0.8 kcal/mol (Figures 3 and 4). Moreover, Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) analysis revealed a substantial binding free energy of -83.28 kcal/mol, indicative of a highly favorable and stable binding affinity between the fusion protein and the receptor subunits. Table 3 illustrates the estimated per-residue energy contributions that are crucial for complex formation.
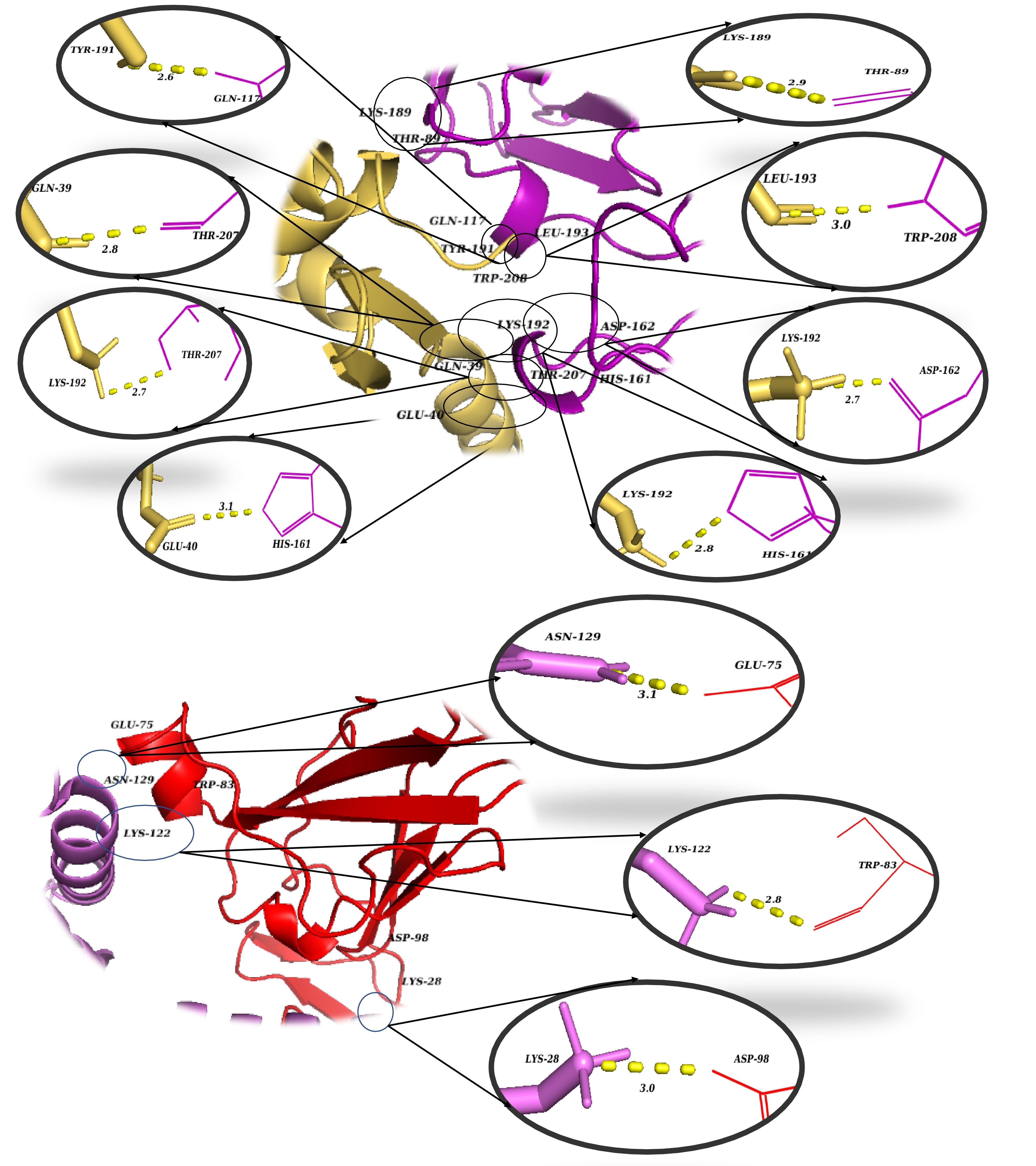
3D visualization of the critical intermolecular interactions at the binding interfaces of the docked complex. Magnified spatial views detail the specific amino acid contacts that stabilize the protein-receptor complex. The top panel illustrates the interaction network between the magainin 2–IL-24 fusion protein (yellow) and the IL-22R1 receptor subunit (purple/magenta). The bottom panel depicts the distinct residue interactions between the fusion protein (violet) and the IL-20R2 receptor subunit (red). Enlarged circular insets provide atomic-level views of individual non-covalent contacts, with yellow dashed lines representing key stabilizing forces such as hydrogen bonds and salt bridges. The numerical values adjacent to the dashed lines denote the corresponding intermolecular bond distances in Ångströms (Å).
Five essential residues contributions to per-residue energy, were determined by Hawk Dock MM/GBSA analysis for an equilibrium simulation trajectory of the structural complex.
| Structure | Rank | Residue | Binding free energy |
|---|---|---|---|
| IL-24 heterodimer receptor | 1 | C-TRP208 | -9.5 |
| 2 | B-ASP98 | -5.12 | |
| 3 | C-TYR60 | -4.4 | |
| 4 | B-TYR74 | -3.93 | |
| 5 | C-ASP162 | -3.64 | |
| Magainin 2-IL 24 fusion protein | 1 | A-LYS28 | -5.02 |
| 2 | A-TRP57 | -3.88 | |
| 3 | A-TYR191 | -3.77 | |
| 4 | A-ASN129 | -2.71 | |
| 5 | A-GLN39 | -2.34 |
Molecular dynamics simulation
The The MD simulation graphs for the docked complex provide critical insights into the stability and dynamic behavior of the protein-receptor complex over a 100 ns trajectory. The RMSD graph (Figure 5A) illustrates the structural stability of the protein during the simulation. Initially, a sharp increase in RMSD is observed, which stabilizes around 20 ns and remains relatively constant, exhibiting only slight fluctuations around 4-5 Å for the remainder of the simulation. This suggests that the protein has reached a stable conformation following the initial equilibration phase, indicating a robust interaction with the receptor.

Molecular dynamics (MD) simulation trajectory analysis of the docked magainin 2–IL-24 fusion protein-receptor complex over 100 ns.(a) Root-mean-square deviation (RMSD) plot of the protein backbone, demonstrating system equilibration and subsequent structural stabilization at approximately 4–5 Å after 20 ns. (b) Radius of gyration (Rg) plot illustrating the overall structural compactness and folding stability, maintaining a consistent value of ~29 Å throughout the simulation. (c) Root-mean-square fluctuation (RMSF) profile depicting per-residue flexibility. The low baseline fluctuations (~2 Å) indicate overall structural rigidity, while the prominent peak near residue 400 highlights a region of localized loop flexibility.
The Rg (radius of gyration) graph (Figure 5B) delineates the compactness of the protein structure over time. The Rg values demonstrate minor fluctuations around 29 Å throughout the simulation, indicating that the fusion protein maintains a consistent and compact conformation. The lack of significant changes in Rg suggests that the protein does not undergo major conformational shifts and remains stable throughout the simulation.
The RMSF (Root Mean Square Fluctuation) graph (Figure 5C) characterizes the flexibility of individual residues within the protein. Most residues exhibit relatively low fluctuations (approximately 2 Å), indicating that the majority of the protein remains stable. However, a noticeable peak is evident around residue 400, suggesting a region of increased flexibility. This could indicate the presence of a loop or unstructured region within the protein that contributes to localized flexibility, which might be functionally significant for the protein’s interaction with the receptor.
Overall, the MD simulation results indicate that the magainin 2-IL24 fusion protein forms a stable complex with its cognate receptor, maintaining structural integrity and consistent interactions throughout the simulation period. The observed stability and minor fluctuations reinforce the potential efficacy of this fusion protein as a therapeutic agent for targeting breast cancer cells.
DISCUSSION
Breast cancer is the most commonly diagnosed cancer worldwide, with 2.3 million new cases and 685,000 deaths reported in 2020, representing a leading cause of female cancer mortality38. Current treatments, including surgery, radiotherapy, chemotherapy, endocrine, and molecular therapies—though effective—are often associated with severe side effects such as cardiotoxicity and systemic toxicity. Moreover, the emergence of multidrug resistance underscores the urgent need for safer and more targeted therapeutic alternatives.
Previously, various types of fusion proteins have demonstrated their potential as innovative therapeutic agents in oncology, particularly for the treatment of breast cancer. Our findings are consistent with previous studies demonstrating that rationally designed fusion proteins can enhance cancer-targeted cytotoxicity while maintaining structural stability. Similar to our construct, chimeric proteins based on Trastuzumab fused with toxins such as Pseudomonas exotoxin A have shown strong receptor-binding affinity in docking analyses and favorable structural validation by Ramachandran assessment, paralleling our results of stable receptor interaction and high-quality 3D modeling39. Likewise, the BENTEC multi-bacteriocin fusion protein demonstrated that the incorporation of membrane-active peptides significantly enhances apoptosis induction in cancer cells, supporting our strategy of integrating the membrane-lytic peptide magainin-2 to potentiate IL-24–mediated tumor targeting40. Additionally, studies on pore-forming toxins such as RTX-A have confirmed that computational optimization can produce stable, soluble, and biologically active fusion constructs, aligning with our molecular dynamics data that demonstrate a stable docked complex41. Rehman et al. (2024) designed a chimeric protein by fusing a cell-penetrating peptide with Interleukin-24 (IL-24), resulting in a bifunctional peptide with enhanced anti-tumor activity, as confirmed through in silico methods42. Similarly, Aslam et al. demonstrated the successful application of azurin protein against breast cancer, validating its cytotoxic effects both in silico and in vitro43. Additionally, Rehman et al. engineered melittin-IL-2444 and IL 24-LK619 fusion proteins, showing promising results regarding stability, functionality, and potential therapeutic application. Meanwhile, Pourhadi et al.45 and Ghavimi et al.46 explored the fusion of IL-24 with BR2 and P28 peptides, respectively, emphasizing the importance of linker selection for maintaining the biological functions of the fused moieties. Furthermore, Moghadam et al.47 and Keshtvarz et al.48 introduced novel fusion proteins combining tumor-targeting peptides with toxin subunits, aiming to enhance specificity and efficacy in targeting cancer cells. In the context of breast cancer, Khalid et al.49, Rehman et al.50, and Qureshi et al.51 designed leptulipin and p28 fusion, azurin-BR2 fusion, and IL24-p20 chimeric proteins, respectively, through computational approaches.
In this study, a novel fusion protein combining IL-24 with magainin 2 was designed and linked via a rigid sequence to create a bifunctional peptide. This fusion was engineered to enhance functionality, with careful consideration given to prevent unwanted interactions. The secondary structure analysis revealed a significant portion of alpha-helical content, indicating strong structural stability, while the presence of beta-sheets and random coils added flexibility and functional diversity. Homology modeling was conducted using AlphaFold2 and I-TASSER (V5.1), with the AlphaFold2 model showing superior accuracy and reliability based on various quality assessments, including the Ramachandran plot, ERRAT score, and Z-score. The physicochemical properties of the fusion protein suggested that it is a stable, soluble, and potentially effective therapeutic agent. Furthermore, the construct was predicted to be non-allergenic, non-antigenic, and non-toxic, making it a promising candidate for therapeutic applications. It was also predicted to be efficiently expressed in E. coli, which is advantageous for downstream applications. The selected docking model exhibited strong binding affinity, as evidenced by favorable scoring metrics, including electrostatic and van der Waals forces, which are key to stabilizing the protein-receptor complex. The presence of multiple salt bridges and hydrogen bonds further reinforced the stability of the interaction, highlighting the potential efficacy of this fusion protein in targeting cancer cells. The substantial number of these non-bonded contacts, particularly at the interfaces between the fusion protein and the IL-22R1 and IL-20R2 subunits, highlights the robustness of the interaction, with specific amino acid residues playing pivotal roles in maintaining the complex's stability. Further analysis of residue interactions revealed crucial amino acid residues involved in stabilizing the protein-protein interface, with MM/GBSA analysis confirming the thermodynamic stability of the complex. The 100 ns molecular dynamics simulation of the magainin 2-IL24 fusion protein complex revealed significant stability, with the complex reaching equilibrium early in the simulation and maintaining a consistent conformation thereafter. The protein's structural compactness remained stable throughout, indicating that the overall structure did not undergo significant changes. Additionally, the majority of the protein residues exhibited rigidity, with only specific loop regions showing increased flexibility. These findings suggest that the protein complex maintained its structural integrity and stability, which is crucial for effective biomolecular interactions.
Several limitations are associated with the current study. First, the accuracy of AlphaFold2 predictions for fusion proteins has not been experimentally validated, and the modeled receptor interactions may not fully reflect in vivo binding specificity. Second, the IL-22R1/IL-20R2 receptor complex is not exclusively expressed on breast cancer cells, indicating potential off-target effects. Third, no experimental controls, such as IL-24 alone docking, were included, and MD simulations were limited to 100 ns, which may not capture long-term dynamics. Finally, predictions of toxicity, antigenicity, and allergenicity rely on computational tools that may generate false positives or negatives. These limitations underscore the necessity for further in vitro and in vivo studies to validate these findings and evaluate cell-type selectivity and off-target effects.
Conclusion
In summary, this in silico study underscores the therapeutic potential of the novel magainin 2–IL-24 fusion peptide as a targeted anticancer agent against breast cancer. Beyond demonstrating structural stability, favorable physicochemical properties, strong receptor binding affinity, and a predicted safety profile, this research establishes a valuable computational framework that can guide future experimental validation and accelerate the rational design of therapeutic fusion proteins, thereby emphasizing the critical role of computational approaches in advancing precision oncology.
Abbreviations
IL: Interleukin; JAK: Janus Kinase; MD: Molecular Dynamics; NAMD: Nanoscale Molecular Dynamics; pI: Isoelectric point; Rg: Radius of gyration; RMSD: Root Mean Square Deviation; RMSF: Root Mean Square Fluctuation; VMD: Visual Molecular Dynamics
Acknowledgments
We acknowledge and express our sincere gratitude to all computational servers, service providers, and software developers whose platforms enabled the successful execution of our in-silico analyses.
Author’s contributions
All authors equally contributed to this work, read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data can be provided on demand from M. Rehman: .
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data).
Competing interests
The authors declare that they have no competing interests.