
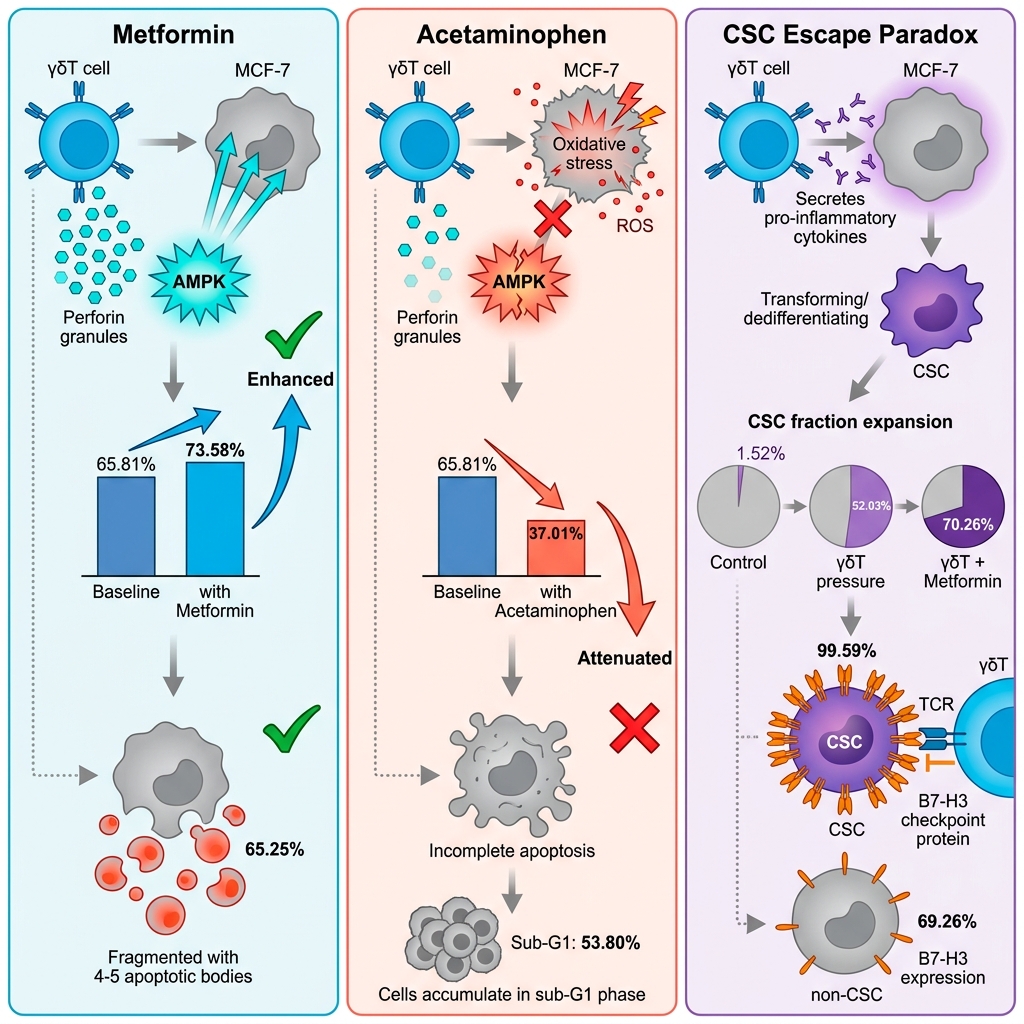

Metformin enhances, whereas acetaminophen attenuates, γδT cell antitumor efficacy and shapes stem-like escape in breast cancer
- VNUHCM-US Stem Cell Institute, University of Science, Viet Nam National University Ho Chi Minh City, Viet Nam
Abstract
Background: Cellular immunotherapies utilizing γδT cells hold immense promise for breast cancer as an MHC-independent approach; however, their clinical efficacy is profoundly dictated by the tumor microenvironment and systemic metabolic contexts. While cancer patients routinely consume widely available pharmacological agents such as Metformin (MET) and Acetaminophen (APAP), their inadvertent modulatory impacts on γδT cell-mediated cytotoxicity and therapy-induced tumor evasion remain fundamentally unexplored.
Methods: We established an in vitro co-culture model utilizing human MCF-7 breast cancer cells and ex vivo expanded primary human γδT cells. Multiparametric flow cytometry and RT-qPCR were employed to interrogate the modulatory effects of MET and APAP on targeted cytotoxicity, apoptotic trajectories, cell cycle dysregulation, the dynamic enrichment of cancer stem cells (CSCs; CD44+CD24-), and the expression profiles of immune checkpoints.
Results: Activated γδT cells exerted robust baseline cytotoxicity, primarily driving target cells into late apoptosis. Strikingly, pharmacological interventions yielded divergent immunomodulatory outcomes. MET synergistically potentiated γδT cell-mediated apoptosis and accelerated tumor clearance. Conversely, APAP profoundly abrogated immune-mediated killing, shifting the death modality toward necrosis and inducing a stalled, pre-lethal sub-G1 cell cycle accumulation. Notably, while γδT cells efficiently eliminated bulk tumor populations, immune-mediated pressure paradoxically enriched the CSC fraction, indicative of aggressive phenotypic plasticity. MET further exacerbated this CSC selection despite maximizing overall cytotoxicity. Furthermore, the surviving stem-like fraction exhibited a marked post-transcriptional upregulation of the immune checkpoint B7-H3 (CD276), revealing a potent adaptive resistance mechanism.
Conclusions: Our findings unveil a critical translational dichotomy: MET acts as a synergistic immunometabolic adjuvant, whereas concurrent APAP exposure may inadvertently compromise γδT cell efficacy. Furthermore, therapy-induced CSC enrichment coupled with B7-H3 upregulation highlights a distinct stem-like immune evasion strategy, underscoring the necessity of combining cellular therapies with B7-H3 blockade to eradicate residual tumor plasticity and prevent relapse.
Introduction
Despite considerable advancements in targeted therapies, breast cancer remains a formidable clinical challenge. This is largely driven by profound intratumoral heterogeneity and adaptive therapeutic resistance, which inevitably lead to disease recurrence 1,2. Central to this refractory behavior is a dynamic subpopulation of tumor cells known as cancer stem cells (CSCs). Rather than existing merely as a static hierarchical apex, breast cancer cells exhibit significant phenotypic plasticity, allowing them to dynamically transition toward dedifferentiated, stem-like states in response to therapeutic stress. Endowed with enhanced survival capacities and intrinsic immune-evasive properties, this plastic subpopulation serves as the primary architect of treatment failure 3,4,5.
In the rapidly evolving landscape of cellular immunotherapy, unconventional γδT cells have emerged as highly attractive innate-like effectors 6,7,8,9. Unlike conventional αβT cells, γδT cells recognize and eradicate transformed cells in a major histocompatibility complex (MHC)-independent manner, positioning them as ideal candidates to target MHC-deficient CSCs 10,11,12. Despite their robust baseline cytotoxicity, the clinical translation of γδT cell therapies is frequently compromised by the metabolically hostile tumor microenvironment (TME) 13,14,15. The anti-tumor efficacy of cytotoxic lymphocytes is inextricably linked to their metabolic fitness, rendering them highly susceptible to localized metabolic perturbations and redox imbalances within the tumor niche.
Crucially, the interaction between immune effectors and tumor cells does not occur in a pharmacological vacuum. In real-world clinical settings, cancer patients routinely consume off-target, non-oncological medications that inadvertently alter systemic and localized metabolic networks. Among these, Metformin (MET), a first-line antidiabetic drug, has garnered intense interest as an archetypal immunometabolic adjuvant. By activating the AMPK signaling axis, MET induces metabolic stress in tumor cells while potentially reinforcing the anti-tumor functions of cytotoxic lymphocytes 16,17,18,19,20,21,22. Recent evidence further indicates that MET can enhance γδT cell function through HIF-1α–mediated dual metabolic reprogramming, reinforcing glycolytic fitness and effector responses under hypoxic conditions 23. Conversely, Acetaminophen (APAP), arguably the most widely consumed over-the-counter analgesic in oncology, induces profound intracellular redox imbalances, glutathione depletion, and mitochondrial dysfunction at elevated local concentrations 24,25,26,27,28,29,30,31,32. Despite the ubiquitous clinical presence of these agents, a fundamental translational blind spot remains: their inadvertent modulatory impacts on γδT cell-mediated cytotoxicity. It is critical to understand whether such concomitant metabolic interventions potentiate immune clearance or paradoxically trigger therapy-induced tumor escape.
Furthermore, it is increasingly recognized that immune-mediated cytotoxicity imposes a profound selective pressure, which may force surviving tumor cells to undergo phenotypic dedifferentiation to escape destruction. In response to this immune-metabolic stress, tumors frequently upregulate non-classical immune checkpoints, such as B7-H3 (CD276) – a molecule strongly implicated in CSC enrichment, immune evasion, and adaptive resistance 33,34,35,36. However, the precise mechanisms by which exogenous metabolic stressors like MET and APAP dictate this therapy-induced CSC enrichment and coordinate the adaptive expression of B7-H3 during γδT cell attack remain entirely unknown.
To bridge this translational knowledge gap, the present study utilizes MET and APAP as prototypical modulators of targeted metabolic and redox stress to interrogate their divergent impacts on primary human γδT cell anti-tumor efficacy against MCF-7 breast cancer cells. By employing an in vitro co-culture paradigm, we comprehensively map (i) the opposing trajectories of immune-mediated apoptosis and cell cycle dysregulation, (ii) the dynamic therapy-induced enrichment of the CSC (CD44CD24) subpopulation, and (iii) the adaptive expression profiles of key immune checkpoints, notably the post-transcriptional deployment of B7-H3. Our findings seek to define the metabolic contexts that either synergize with or sabotage γδT cell immunotherapy, providing crucial insights into therapy-induced phenotypic plasticity.
Materials and Methods
Cell culture and reagents
The human luminal A breast cancer cell line MCF-7 (ATCC) was maintained in RPMI 1640 medium (Gibco, Cat# 61870-036) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Cat# A5256701) and 1X antibiotic-antimycotic solution (Gibco, Cat# 15240-062). Cultures were incubated at 37°C in a humidified 5% CO atmosphere, with medium replenished every 48 h. Subculturing was performed utilizing TrypLE Express (Gibco, Cat# 12604021) upon reaching 80-90% confluence. Routine testing was performed to ensure cell lines were free of Mycoplasma contamination prior to functional assays. APAP (Sigma-Aldrich, Cat# A7085) was dissolved in DMSO to generate a 5 M stock, ensuring the final DMSO concentration in all experimental conditions strictly remained ≤ 1% (v/v) to circumvent vehicle-induced toxicity. MET (Sigma-Aldrich, Cat# 317240) was prepared as a 1 M stock in sterile nuclease-free water.
Ex Vivo expansion and phenotypic validation of primary γδT cells
Isolation and expansion
Peripheral blood mononuclear cells (PBMCs) were isolated via standard density-gradient centrifugation (1500 × g, 15 min). For specific γδT cell activation, PBMCs were initially cultured in GDTCult D1-3 medium (Regenmedlab, Cat# 354) for 48 h. The non-adherent fraction was subsequently transferred to gas-permeable culture bags and robustly expanded in GDTCult D3-21 medium, supplemented every 3 days. Post-21 days of expansion, the effector cells were harvested for downstream immunological interrogations.
Flow cytometric quality control
Prior to functional co-cultures, the purity of the expanded effector population was rigorously verified. Cells (0.5-2 x 10) were stained with PerCP-Cy5.5-conjugated Mouse Anti-Human TCR γδ (BD Pharmingen, Cat# 564157). Phenotypic acquisition was performed using a BD FACS Melody flow cytometer (BD Biosciences). Only expanded products demonstrating a stringent purity threshold of ≥ 50% TCR γδ cells were classified as functionally competent and utilized for subsequent assays.
Pharmacological modulation and metabolic stress modeling
To establish in vitro models of acute, targeted metabolic and redox stress, the half-maximal inhibitory concentrations (IC) of APAP and MET on MCF-7 cells were predetermined. Cells (2,000 cells/well) were exposed to dose-escalations of APAP (0–100,000 µM) or MET (0–20,000 µM). Viability was quantified at 24, 48, and 72 h via the AlamarBlue assay (Thermo Fisher, Cat# DAL1025). Based on nonlinear regression analyses, stress-inducing concentrations corresponding to their 48-h kinetic profiles (10 mM APAP and 20 mM MET) were selected for downstream immune co-cultures to evaluate modulatory effects without inducing absolute target cell ablation (The IC₅₀ values were previously determined by our team). These concentrations were intentionally selected to model acute, localized metabolic or redox stress conditions that may occur within TME, rather than systemic plasma levels.
Multiparametric assessment of immune-mediated cytotoxicity and cell cycle dysregulation
Co-culture paradigm
Target MCF-7 cells were seeded in T75 flasks and allowed to adhere for 48 h. Expanded primary γδT cells were subsequently introduced at a predefined effector-to-target (E:T) ratio of 20:1. Co-cultures were maintained for 48 h either in basal media or under pharmacological stress (10 mM APAP or 20 mM MET).
Apoptotic trajectories and viability mapping
Following immune challenge, overall target cell death was quantified utilizing 7-Aminoactinomycin D (7-AAD; BD Pharmingen, Cat# 559925) exclusion staining. To precisely delineate early/late apoptotic trajectories and necrotic shifts, cells were dual-stained with Annexin V-FITC and Propidium Iodide (PI) (BD Pharmingen, Cat# 556547).
Cell cycle profiling
Cell cycle progression and sub-G1 pre-lethal DNA fragmentation were quantified using PI/RNase Staining Buffer (BD Pharmingen, Cat# 550825) following overnight fixation in 70% ice-cold ethanol at -20°C. Data were mathematically modeled using FlowJo v10.8 Software (BD Biosciences).
Crucially, for all co-culture flow cytometric analyses, target MCF-7 cells and effector γδT cells were computationally discriminated based on their distinct Forward Scatter (FSC) and Side Scatter (SSC) profiles. Sequential hierarchical gating strategies were strictly applied to exclude cellular debris and eliminate doublets (FSC-H vs. FSC-A) to ensure single-cell analytical fidelity.
Phenotypic plasticity analysis and cancer stem cell (CSC) sorting
To induce stem-like enrichment via a serum-deprivation stress model, MCF-7 cells were cultured in media supplemented with varying FBS concentrations (10%, 2%, and 0.4%). To delineate the CSC subpopulation, harvested cells were immunolabeled at 4°C in the dark with PE-conjugated Anti-Human CD44 (IM7) and FITC-conjugated Anti-Human CD24 (M1/69) monoclonal antibodies (BD Biosciences). The stem-like subset was phenotypically defined as CD44CD24. Fluorescence-activated cell sorting (FACS) was performed using the BD FACS Melody system to isolate high-purity CSC and non-CSC (CD44CD24 and CD44CD24) fractions, which were subsequently subjected to γδT cell co-culture across escalating E:T ratios (0:1 to 50:1) to profile differential immune susceptibility.
Transcriptional and transmembrane immune checkpoint profiling
Total RNA from sorted fractions (1-10 x 10 cells) was extracted utilizing the easy-BLUE™ Total RNA Extraction Kit (iNtRON Biotechnology, Cat# 17061). RNA concentration and purity (A260/280 ratio) were verified via a NanoDrop spectrophotometer. The mRNA expression profiles of key immune modulators (CD95, CD95L, and B7-H3) were quantified via one-step RT-qPCR utilizing the Luna Universal One-Step RT-qPCR Kit (NEB, Cat# E3005) on a LightCycler 480 II (Roche). Relative target gene expression was mathematically derived using the 2 method, with GAPDH acting as the endogenous internal reference (primer sequences detailed in Supplementary Table S1). Parallelly, post-transcriptional surface expression of B7-H3 and CD95L on CSCs versus non-CSCs was validated via flow cytometry.
Statistical comparisons
All empirical quantitative data are presented as the Mean ± Standard Deviation (SD) of at least three independent biological replicates (n = 3). Statistical comparisons were executed using GraphPad Prism version 10.4.1 (GraphPad Software, San Diego, CA). Data normality was assessed using the Shapiro–Wilk test prior to parametric analyses. Exact p-values were reported whenever applicable, whereas threshold notation (*p < 0.05, **p < 0.01, etc.) was retained in graphical summaries for visual clarity. For strict two-group comparisons, an unpaired two-tailed Student’s t-test with Welch’s correction was applied. For multi-group comparisons encompassing 3 experimental conditions (e.g., assessing diverse treatment conditions against a control), a One-Way Analysis of Variance (ANOVA) was performed, followed by Tukey’s or Dunnett’s multiple comparisons post-hoc tests to strictly control the family-wise Type I error rate. A predefined alpha level of p < 0.05 was established for statistical significance.
Results
Metformin synergizes with γδT cell cytotoxicity, whereas acetaminophen attenuates target cell clearance
To establish the baseline anti-tumor efficacy of expanded effector cells, primary human γδT cells (routinely exhibiting ≥ 89.9% TCRγδ purity; Figure S1) were co-cultured with MCF-7 breast cancer cells. Morphological assessments via phase-contrast microscopy revealed that γδT cell engagement induced profound target cell destruction, characterized by marked reductions in cell density, robust cellular flattening, and the formation of pseudopodia-like protrusions (Figure 1). Quantitative 7-AAD exclusion assays corroborated these observations, demonstrating a striking increase in MCF-7 cell death following γδT cell co-culture (65.81 ± 0.64%) compared to the spontaneous basal turnover in untreated controls (7.00 ± 0.27%, p < 0.0001) (Figure 2).

Phase-contrast microscopy of MCF-7 cell morphology under γδT cell co-culture and metabolic stress. At 48 h, untreated MCF-7 cells displayed normal epithelial morphology and reached near confluence. Co-culture with γδ T cells (E:T = 20:1) decreased cell density and triggered morphological changes, including cell flattening and pseudopodia formation. Concurrent treatment with APAP amplified these effects, markedly reducing cell density and increasing cellular elongation, mimicking the morphology of APAP-only treated cells. The MET co-culture group exhibited comparable morphological alterations, though MET monotherapy resulted in less severe density reduction than APAP.
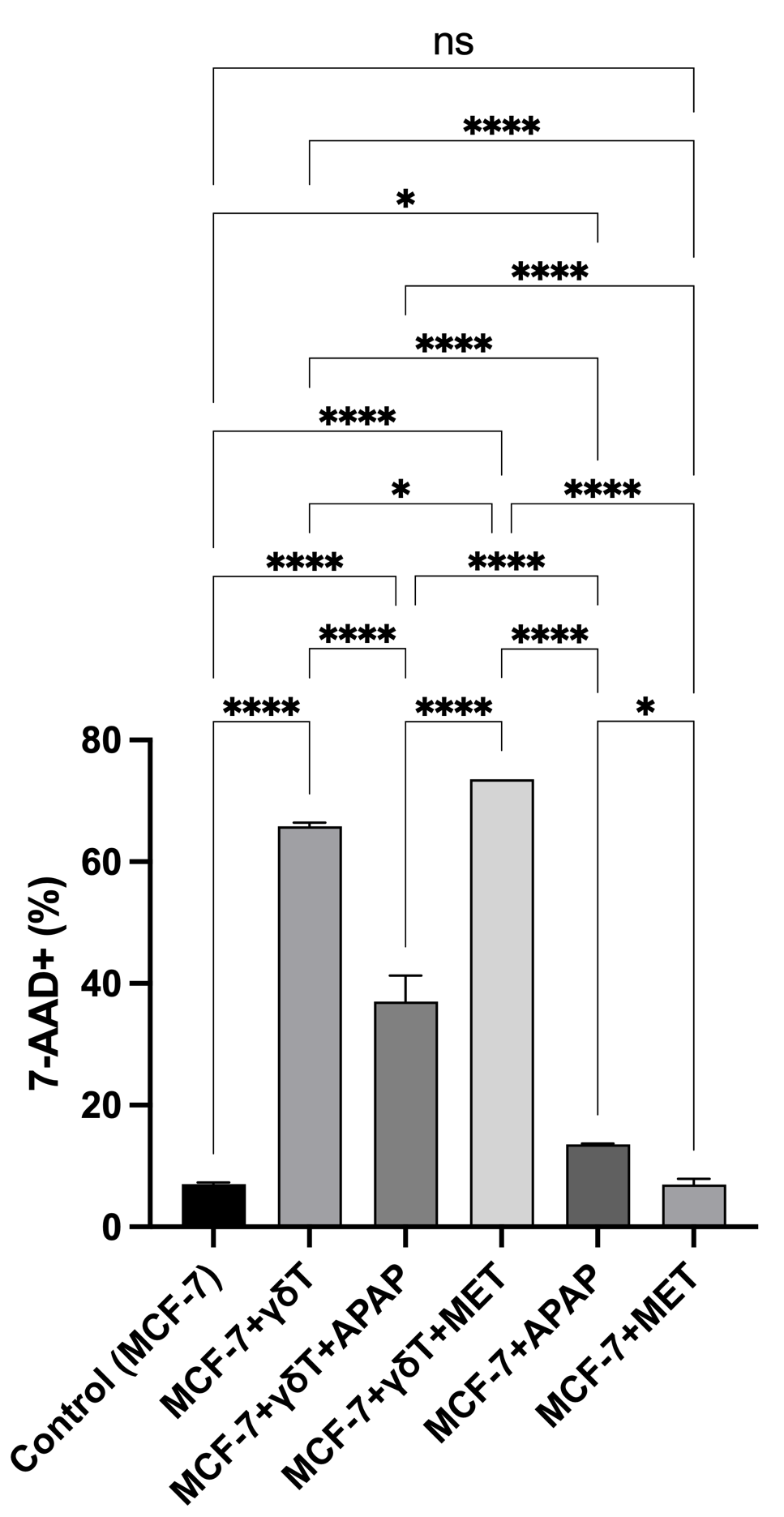
Assessment of MCF-7 cell death during co-culture with γδ T cells, with or without APAP and MET. 7-AAD staining showed that co-culture with γδ T cells (E:T = 20:1) markedly increased MCF-7 cell death compared with the control. APAP (10 mM) significantly reduced the cytotoxic activity of γδ T cells, whereas MET increased the effect. Treatment with APAP or MET alone moderately increased MCF-7 cell death relative to spontaneous cell death after 48 h of culture.
Crucially, pharmacological interventions completely bifurcated this immune-mediated clearance. The introduction of MET (20 mM) synergistically augmented γδT cell cytotoxicity, driving overall target cell death to 73.58% (p < 0.05 vs. γδT alone). Conversely, concurrent exposure to APAP (10 mM) profoundly abrogated immune efficacy, rescuing a significant proportion of MCF-7 cells and plummeting the death rate to 37.01 ± 4.30% (p < 0.0001 vs. γδT alone). Importantly, monotherapies of APAP or MET exerted negligible to modest direct cytotoxicity on MCF-7 cells (13.60 ± 0.11% and 6.97 ± 0.91%, respectively), confirming that their primary impact lies in the modulation of the immune-tumor interaction rather than direct generalized toxicity.
Divergent modulation of apoptotic trajectories and cell cycle arrest by metabolic stressors
To elucidate the mechanistic underpinnings of these opposing cytotoxic outcomes, we mapped the cell death modalities and cell cycle dynamics via Annexin V/PI profiling and PI/RNase DNA content analysis. In the absence of pharmacological stress, γδT cells predominantly eliminated MCF-7 targets via late apoptosis (25.90 ± 0.85% vs. 11.70 ± 0.17% in control, p < 0.0001), accompanied by a significant accumulation of cells in the sub-G1 phase (16.70 ± 5.35%, p < 0.0001) and a concurrent collapse of the G2 compartment (Figures 3 and 4).
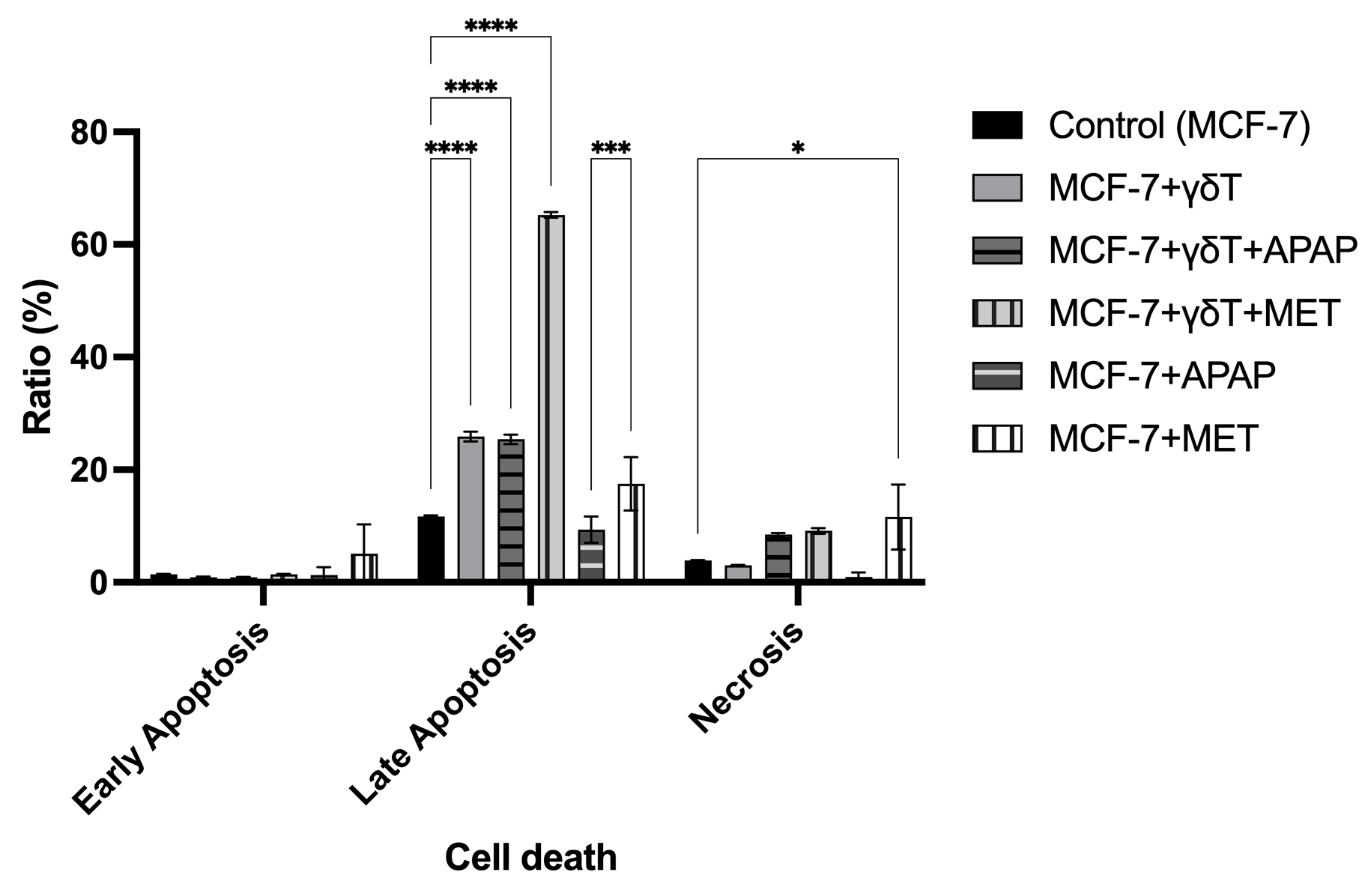
Cell death phenotypes of MCF-7 cells during co-culture with γδ T cells, with or without APAP and MET. Annexin V/PI analysis showed that γδ T cells significantly increased late apoptosis in MCF-7 cells (25.90 ± 0.85% vs. 11.70 ± 0.17% in control, p < 0.0001). Co-treatment with MET further enhanced late apoptosis to 65.25 ± 0.50% (p < 0.0001), whereas APAP did not further increase apoptosis but significantly promoted necrotic cell death (8.52 ± 0.28%, p < 0.05). APAP alone caused only modest apoptotic changes, whereas MET alone induced both apoptotic and necrotic responses, particularly increasing late apoptosis and necrosis (n = 3).
Mirroring the 7-AAD survival data, MET and APAP orchestrated diametrically opposed apoptotic trajectories. MET robustly potentiated immune-mediated late apoptosis, driving the apoptotic fraction to an overwhelming 65.25 ± 0.50% (p < 0.0001). In stark contrast, APAP failed to enhance γδT cell-induced apoptosis (25.40 ± 0.82%, p > 0.05 vs. γδT alone), but instead promoted a shift toward necrotic cell death (8.52 ± 0.28%, p < 0.05). Most strikingly, cell cycle analysis revealed that the addition of APAP to the co-culture induced a massive, stalled accumulation of target cells in the sub-G1 phase (53.80 ± 3.04%, p < 0.0001) with total ablation of the G2 mitotic population (0.00%). This extreme sub-G1 accumulation, together with the reduced terminal membrane permeability detected by 7-AAD staining, suggests that APAP may interfere with the effective completion of γδT cell-induced apoptosis. Rather than representing definitive apoptotic execution, this pattern may reflect incomplete or stalled apoptotic progression associated with extensive DNA fragmentation but limited terminal cell death.
γδT cell-mediated immune pressure drives phenotypic plasticity and CSC enrichment
We next investigated whether this immune-metabolic stress landscape dynamically alters the hierarchical composition of the tumor, specifically the enrichment of Cancer Stem Cells (CSCs; functionally defined here as CD44CD24). We first validated that severe physiological stress (0.4% FBS starvation) robustly enriched the CSC phenotype to nearly 80% (Figures 5 and 6), confirming the profound plasticity of MCF-7 cells.
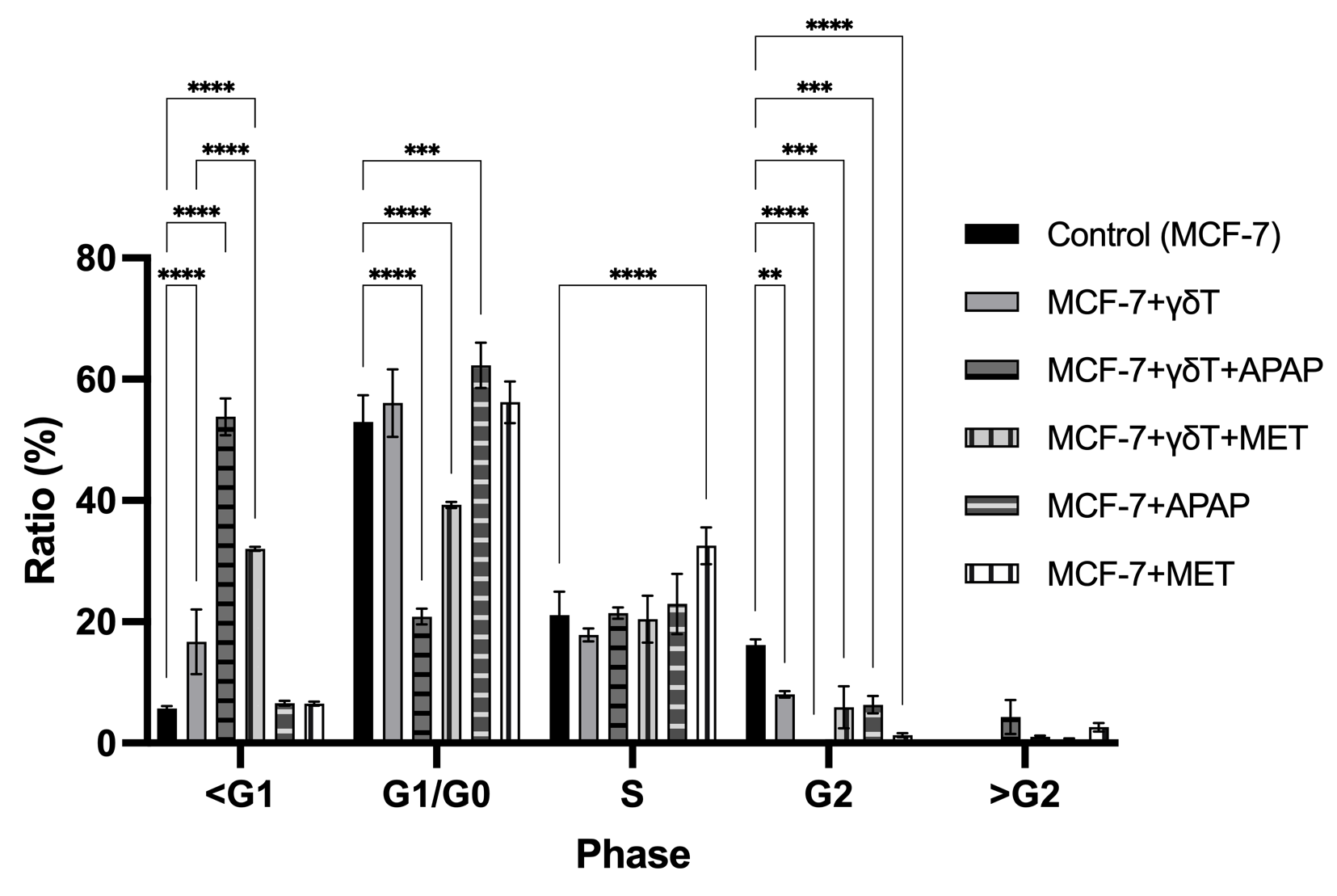
Cell cycle analysis of MCF-7 cells during co-culture with γδ T cells, with or without APAP and MET. Cell cycle analysis showed that co-culture with γδ T cells significantly increased the sub-G1 population of MCF-7 cells (5.70 ± 0.40% to 16.70 ± 5.35%, p < 0.0001) and reduced the G2 fraction (16.17 ± 0.93% to 8.05 ± 0.52%, p < 0.01). The combination of γδ T cells and APAP produced the most pronounced effect, markedly increasing the sub-G1 population to 53.80 ± 3.04% (p < 0.0001) and nearly abolishing the G2 phase. γδ T cells combined with MET moderately increased the sub-G1 fraction (32.03 ± 0.35%, p < 0.0001 vs. control) and reduced G2 cells (p < 0.001). APAP alone induced mild G1/G0 accumulation (62.30 ± 3.73%, p < 0.001) with reduced G2 (p < 0.001), whereas MET alone slightly increased the S-phase population (32.53 ± 3.03%, p < 0.001) and decreased G2 cells (p < 0.0001). These results suggest that APAP strongly enhances pre-lethal DNA fragmentation, whereas MET predominantly promotes apoptotic progression
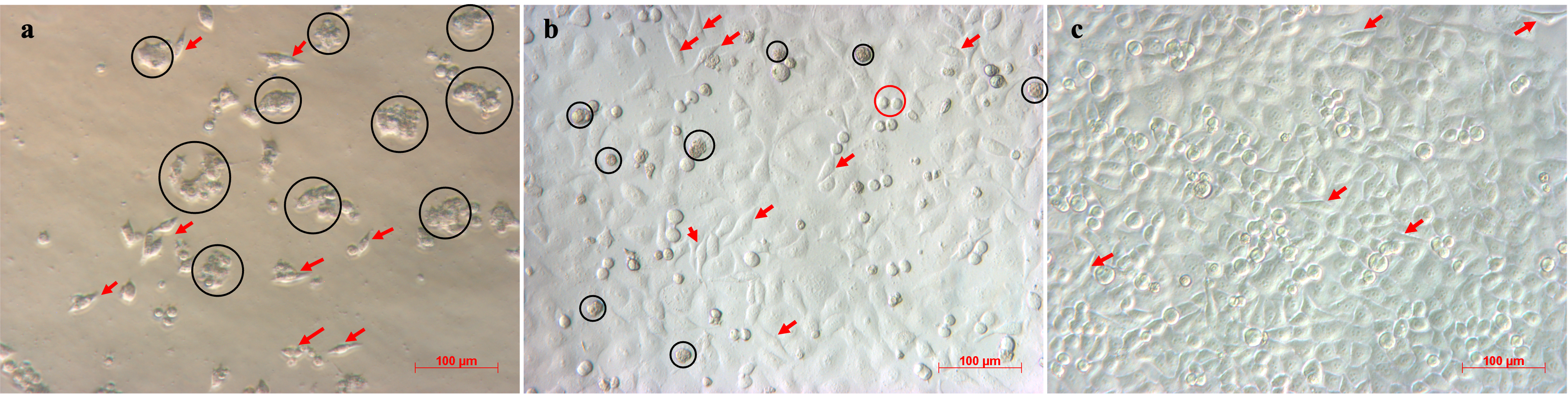
Images of MCF-7 cells cultured in medium with three different FBS concentrations: (a) 0.4% FBS, (b) 2% FBS, (c) 10% FBS. MCF-7 cells cultured under decreasing FBS concentrations showed reduced proliferation and altered morphology. Cells in 10% FBS displayed high proliferative activity and typical epithelial morphology, whereas 2% FBS resulted in slower growth, increased cell elongation, and higher cell death. In contrast, 0.4% FBS markedly inhibited proliferation, induced extensive cell death, and promoted a fibroblast-like morphology, with cells nearly ceasing growth after 3–5 days.
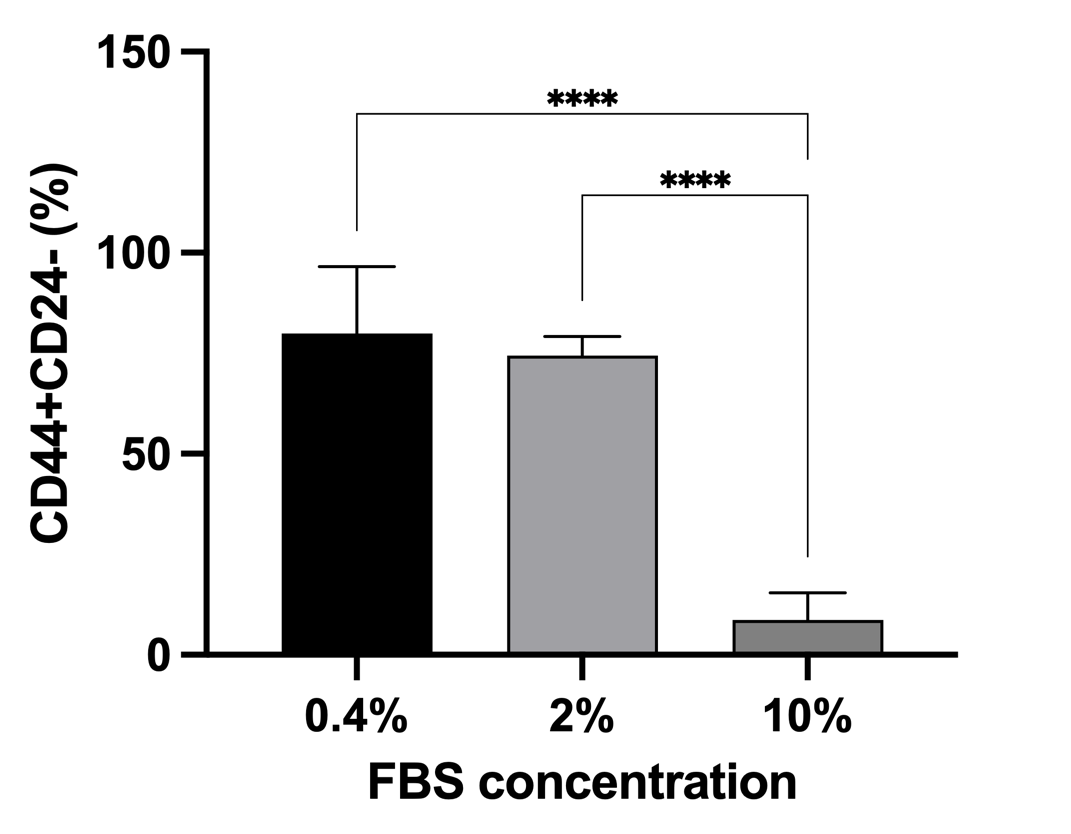
MCF-7 cancer stem cells enrichment in different serum-deprived conditions. FACS analysis of CD44⁺CD24⁻ cells showed that serum deprivation increased the CSC fraction in MCF-7 cultures. Both 0.4% and 2% FBS conditions significantly enriched CSCs compared to 10% FBS (p < 0.0001), with the highest proportion observed at 0.4% FBS. Despite similar CSC enrichment between 0.4% and 2% FBS, improved cell growth under 2% FBS suggests it as an optimal condition for CSC enrichment.
To determine if CSC enrichment is driven by intrinsic resistance or acquired plasticity, we sorted high-purity CSCs and non-CSCs and exposed them independently to γδT cells. Strikingly, both populations exhibited near-identical susceptibility to immune-mediated clearance across all tested E:T ratios (Figure 7), proving that baseline CSCs are not inherently resistant to γδT cells.
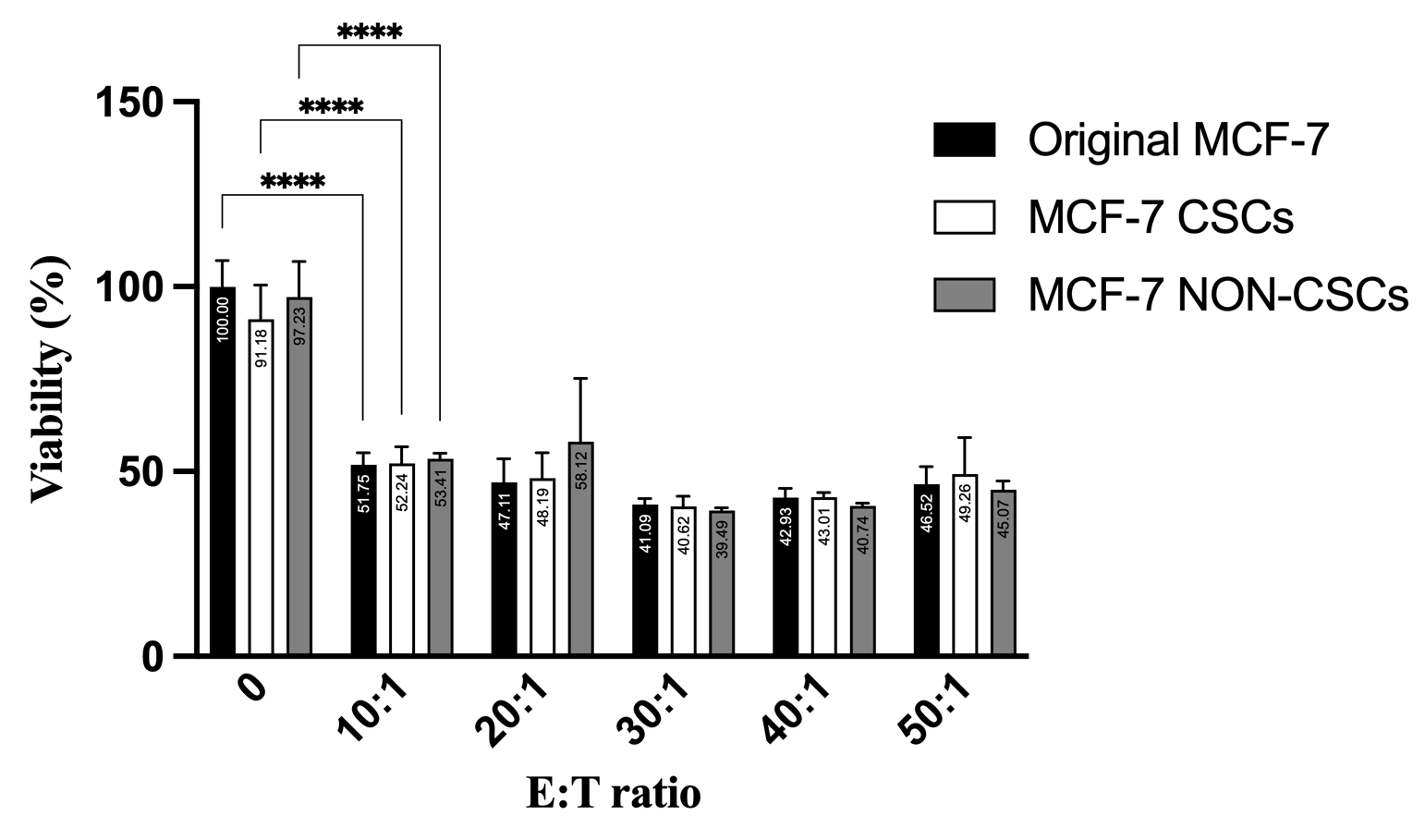
Proliferation capacity of MCF-7 origin, CSCs, and non-CSCs during co-culture with γδ T cells across different E:T ratios. γδ T cells co-culture significantly inhibited the proliferation of MCF-7 cells in a dose-dependent manner starting at E:T = 10:1. Comparable reductions in proliferation were observed across total MCF-7 cells, CSCs (CD44⁺CD24⁻), and non-CSCs at all tested E:T ratios, with no significant differences in susceptibility among the subpopulations
However, within the bulk tumor co-culture, the selective pressure exerted by γδT cells paradoxically triggered a massive phenotypic shift, expanding the surviving CSC fraction from 1.52 ± 0.24% in controls to 52.03 ± 3.02% (p < 0.0001) (Figure 8, Figure S2). This robust enrichment was further exacerbated by MET (70.26%), indicating that maximal immune-metabolic stress forces the surviving cellular remnants into a hyper-stem-like state. Although APAP attenuated this dramatic CSC expansion compared to the γδT-alone group, the CSC fraction remained significantly elevated relative to basal levels (8.85 ± 1.56%, p < 0.01). These data collectively suggest that immune attack dynamically reshapes tumor architecture and may promote the enrichment of a stem-like state as an adaptive survival response.
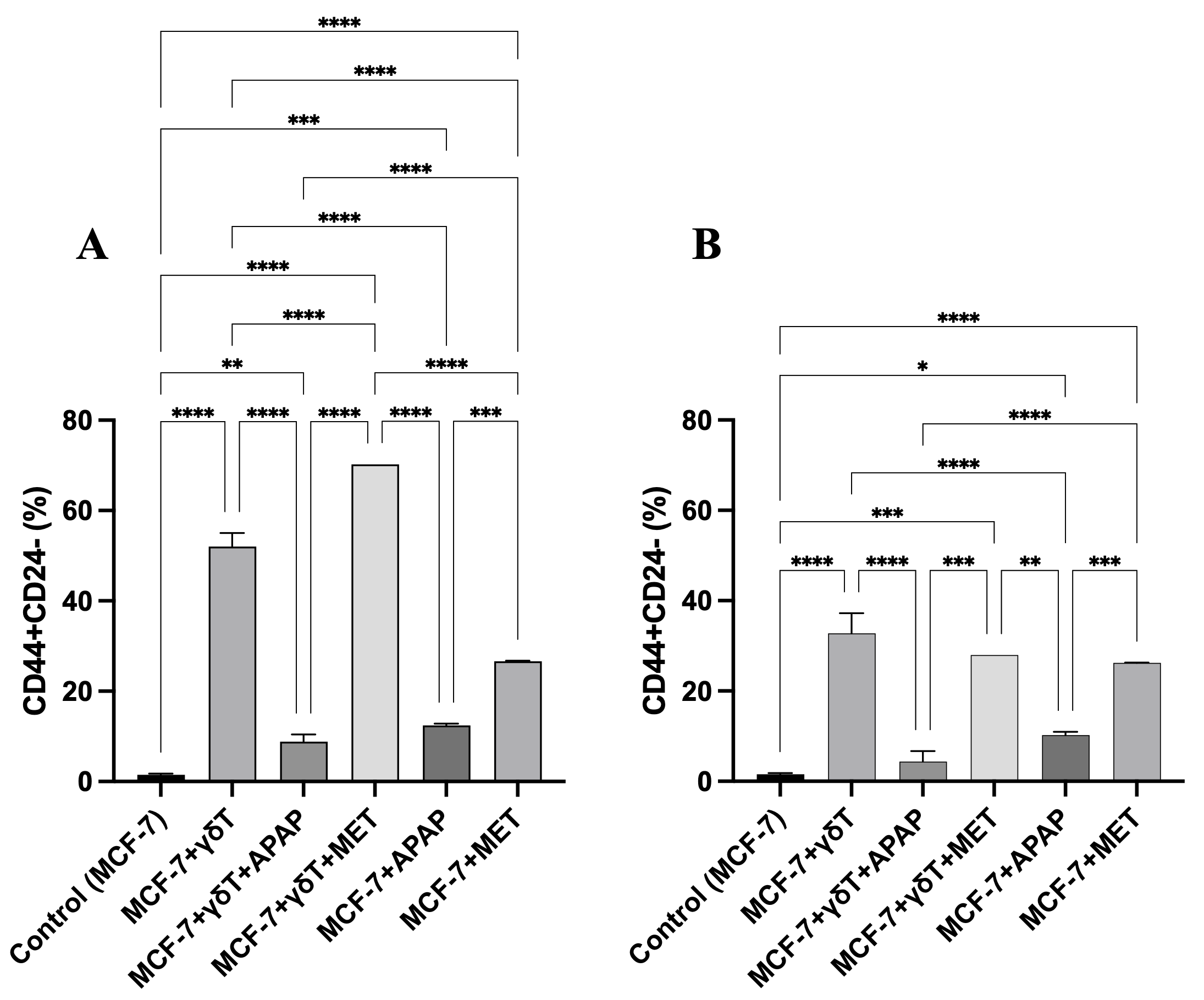
Enrichment of CD44⁺CD24⁻ cancer stem cells (CSCs) in MCF-7 cultures during co-culture with γδ T cells, with or without APAP and MET. (A) Relative CSC proportion within the total cell population (7-AAD⁺/⁻). (B) Relative CSC proportion within the viable cell population only (7-AAD⁻). γδ T-cell treatment markedly increased CSC enrichment compared with the control. APAP reduced CSC enrichment relative to γδ T cells alone but remained above baseline levels, whereas MET further increased CSC accumulation. APAP and MET monotherapies caused only moderate increases in CSC proportion. Minimal 7-AAD-positive CSCs were detected across monotherapies
The Surviving Stem-Like Fraction Exhibits Post-Transcriptional Upregulation of B7-H3 (CD276)
To uncover the molecular shields employed by these enriched CSCs to evade total eradication, we interrogated the expression of canonical (CD95/CD95L) and non-canonical (B7-H3) immune checkpoints. At the transcriptional level, RT-qPCR analysis revealed no statistically significant divergence in mRNA expression for CD95, CD95L, or B7-H3 between sorted CSC and non-CSC populations (p > 0.05), despite a marginal trend toward increased B7-H3 transcripts in CSCs (Figure 9).
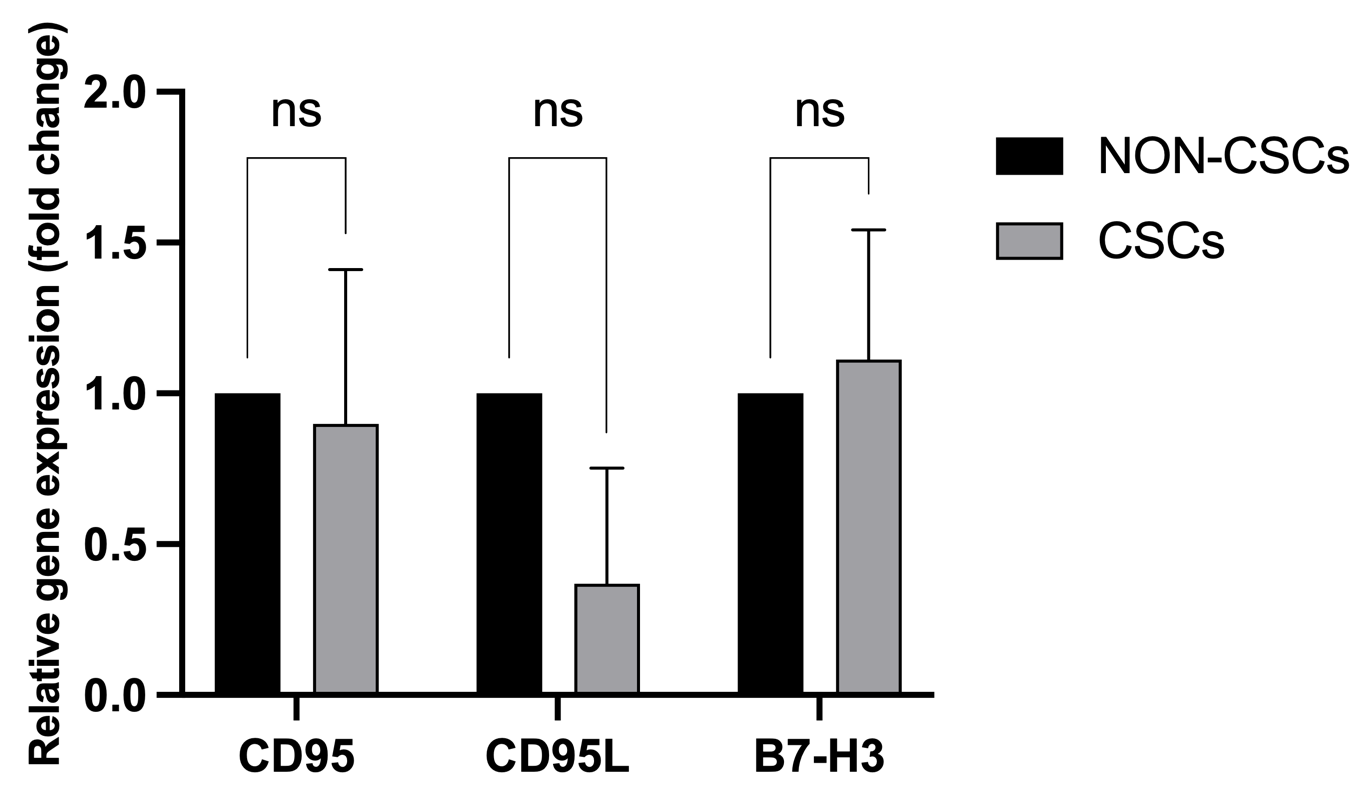
Transcription expression of CD95/CD95L and B7-H3 between MCF-7 CSCs and non-CSCs. mRNA expression analysis of CD95, CD95L, and B7-H3 showed no significant differences between CSCs (CD44⁺CD24⁻) and non-CSCs in MCF-7 cells (p > 0.05), despite a trend toward decreased CD95L and increased B7-H3 expression in CSCs.
Crucially, however, flow cytometric analysis of transmembrane protein expression unveiled a stark post-transcriptional dichotomy. While surface CD95L remained comparable between the subsets, the expression of B7-H3 was significantly upregulated on the surface of CSCs (99.59 ± 0.03%) compared to their non-CSC counterparts (69.26 ± 1.70%, p < 0.05) (Figure 10). This pronounced, preferential deployment of B7-H3 on the CSC membrane highlights a specific, post-transcriptionally regulated adaptive resistance mechanism, equipping the therapy-induced stem-like population with a robust checkpoint shield against sustained γδT cell immunosurveillance.
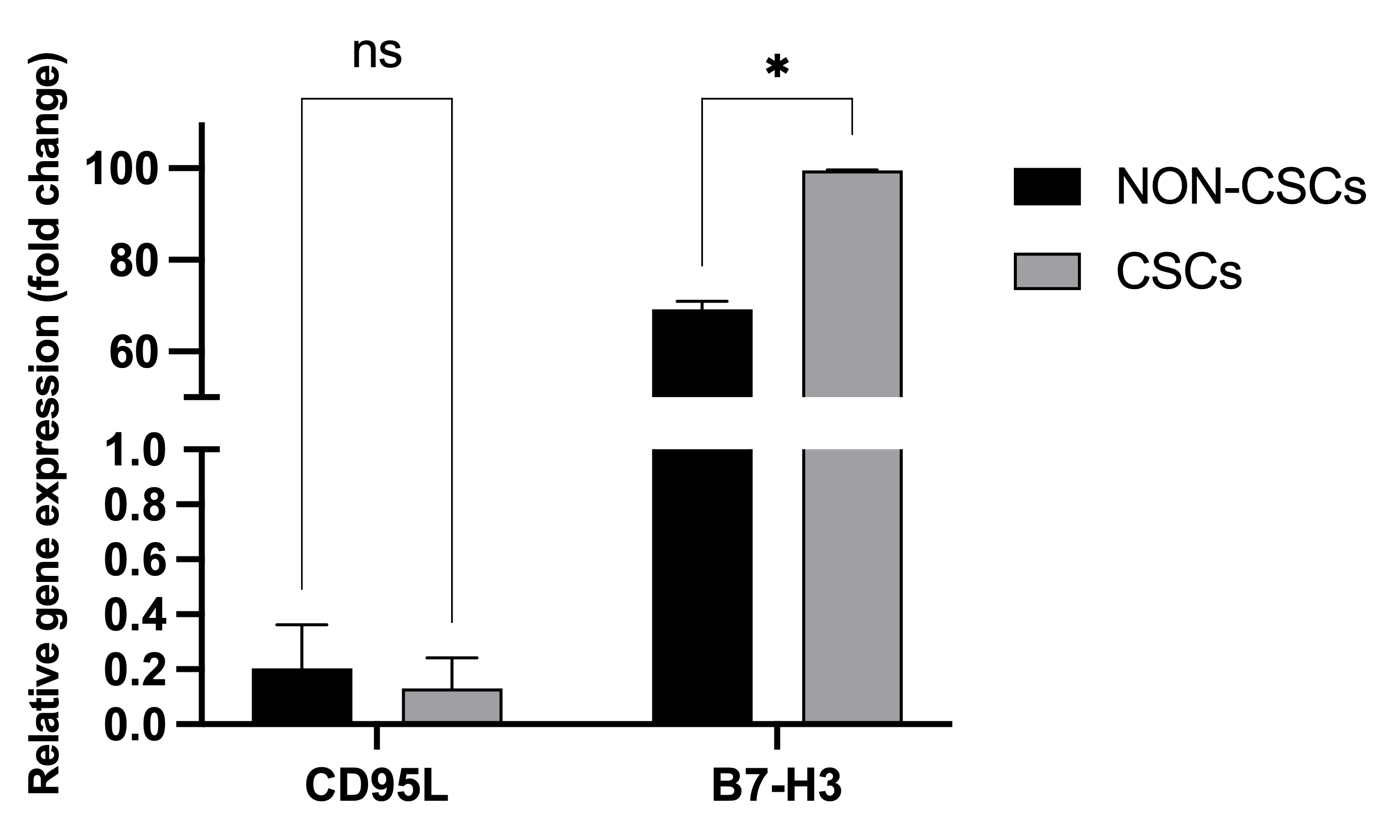
Transmembrane expression of CD95/CD95L and B7-H3 between MCF-7 CSCs and non-CSCs. Flow cytometric analysis showed that B7-H3 protein expression was significantly higher in CSCs (CD44⁺CD24⁻) than in Non-CSCs in MCF-7 cells (99.59 ± 0.03% vs. 69.26 ± 1.70%, *p < 0.05), while CD95L expression showed no significant difference (p > 0.05).
Discussion
The clinical translation of cellular immunotherapies is frequently bottlenecked by the metabolically hostile TME and the off-target effects of concomitant pharmacological agents. Our study unveils a critical translational dichotomy: while MET acts as a potent immunometabolic adjuvant that synergizes with γδT cell-mediated cytotoxicity, APAP – a ubiquitous clinical analgesic – paradoxically sabotages this immune clearance. Crucially, we demonstrate that immune-metabolic pressure dynamically dictates tumor phenotypic plasticity, driving the enrichment of a stem-like subpopulation heavily shielded by the post-transcriptional upregulation of the B7-H3 immune checkpoint.
The profound divergence between MET and APAP in modulating γδT cell efficacy underscores the critical role of targeted metabolic contexts. MET, a canonical AMPK activator, is known to inhibit mitochondrial respiration, thereby inducing a state of metabolic crisis that sensitizes tumor cells to immune-mediated apoptosis 18,19,20,22,37. Our data perfectly align with this, showing a massive potentiation of late apoptosis under MET treatment. Notably, recent work has demonstrated that metformin enhances γδT cell function via HIF-1α–dependent metabolic reprogramming, promoting glycolytic adaptation under hypoxic stress 23. This mechanism may further explain the potentiation of γδT cell cytotoxicity observed in our system, particularly within metabolically constrained tumor-like conditions. In stark contrast, APAP effectively decoupled DNA fragmentation from terminal cell death. Under APAP exposure, MCF-7 cells exhibited a massive, stalled accumulation in the hypodiploid sub-G1 phase with complete G2 ablation, yet successfully evaded membrane rupture (low 7-AAD positivity). We cautiously interpret this phenomenon as a state consistent with “incomplete apoptosis”. This phenomenon is uniquely illuminated in MCF-7 cells, which intrinsically lack caspase-3 and rely on less efficient, non-canonical executioner pathways 38. High-dose APAP induces severe intracellular redox imbalances and glutathione depletion 39,40. Instead of accelerating death, this severe metabolic dysregulation likely disrupts the ATP-dependent execution phase of γδT cell-induced granzyme/death-receptor signaling, stalling the tumor cells in a highly mutagenic, pre-lethal state. Emerging evidence increasingly implicates such sub-lethal apoptotic engagement as a potent driver of genomic instability and adaptive survival, affording tumor cells a temporal window to reprogram their fate 41,42,43. While this phenomenon is particularly evident in caspase-3-deficient MCF-7 cells, we acknowledge that validation in caspase-competent models such as T47D or MDA-MB-231 would be necessary to generalize this mechanism.
Perhaps the most clinically alarming consequence of this incomplete clearance is the robust, therapy-induced enrichment of the Cancer Stem Cell (CD44CD24) fraction. Importantly, our functional sorting assays definitively proved that baseline CSCs are equally susceptible to γδT cell killing as non-CSCs. Therefore, the dramatic expansion of the CSC pool (from ~1.5% to >50%) within the bulk co-culture cannot be fully explained by simple Darwinian selection alone of an intrinsically resistant clone. While selective survival of CSCs may contribute to the observed enrichment, our data suggest that therapy-induced phenotypic plasticity, a rapid dedifferentiation of non-CSCs into a stem-like state, may also play a role. Pro-inflammatory cytokines (e.g., IFN-γ, TNF-α) released during γδT cell attack are known to activate STAT3/NF-κB pathways, driving this exact stemness reprogramming 44,45,46. Notably, MET exacerbated this CSC enrichment despite maximizing overall target clearance, emphasizing the universal biological principle that maximal cytotoxic stress simultaneously provokes maximal phenotypic adaptation among the surviving remnants.
To survive this intense immunosurveillance, these newly enriched CSCs must deploy robust molecular shields. Our data elegantly capture this adaptive response: while transcriptional levels of immune checkpoints remained static, B7-H3 was significantly upregulated at the surface protein level specifically within the CSC compartment. This post-transcriptional deployment of B7-H3 provides a potent non-classical checkpoint barrier, aligning with emerging clinical data identifying B7-H3 as a critical mediator of immune evasion and tumor aggressiveness 33,34,35,36. The reliance on post-transcriptional mechanisms allows for rapid membrane reinforcement without the energetic delay of de novo mRNA synthesis, representing a highly efficient survival strategy under acute immune attack.
We acknowledge that the concentrations of APAP and MET used in this study exceed typical systemic physiological levels. However, these doses were selected to model localized metabolic stress conditions within the tumor microenvironment, where drug accumulation and altered pharmacokinetics may occur. Future studies should incorporate dose titration under clinically relevant concentrations to better define translational applicability.
Limitations and Future Directions
We acknowledge several limitations in the present study. A key limitation of this study is the reliance on a single caspase-3-deficient cell line (MCF-7). While this model is advantageous for uncovering non-canonical apoptotic phenotypes, future validation in caspase-3-competent breast cancer models (e.g., T47D, MDA-MB-231) will be essential to confirm the generalizability of the observed “aborted apoptosis” mechanism. Second, the supraphysiological concentrations of APAP and MET utilized herein serve as robust in vitro tools to model extreme targeted metabolic or redox stress; however, translating these kinetics to systemic in vivo architectures requires careful dose-titration. Third, while our co-culture design focuses on the phenotypic fate of the target tumor cells, the direct pharmacological modulation of the γδT cell immunological synapse by APAP remains an intricate variable. However, even if APAP partially impairs effector cell viability, the profound target-cell sub-G1 arrest strongly indicates that effector cells remain functionally engaged enough to initiate lethal DNA damage, albeit with an aborted terminal execution. Lastly, direct evidence of lineage conversion for tracking the dedifferentiation of cancer cells was not assessed in this study. In future studies, the direct effects of APAP and MET on γδT cell viability and effector function should be independently assessed. Additionally, utilizing complex 3D organoid models and targeted B7-H3 blockade are warranted to fully untangle these bidirectional immunometabolic networks.
Conclusion
This study establishes that concomitant pharmacological metabolic interventions profoundly dictate the trajectory of cellular immunotherapy in breast cancer. Metformin serves as a synergistic adjuvant, accelerating γδT cell-mediated apoptosis, whereas APAP sabotages immune clearance, stalling tumor cells in a highly plastic, pre-lethal state. Crucially, surviving tumor cells escape eradication via dynamic dedifferentiation into a B7-H3-shielded stem-like state. These findings raise significant translational warnings regarding the unmonitored use of over-the-counter analgesics during immunotherapy and strongly advocate for combinatorial strategies targeting B7-H3 to eradicate residual tumor plasticity and prevent therapeutic relapse.
Abbreviations
7-AAD: 7-Aminoactinomycin D; ADCC: Antibody-Dependent Cellular Cytotoxicity; AMPK: AMP-Activated Protein Kinase; APAP: Acetaminophen (Paracetamol); BSA: Bovine Serum Albumin; CSCs: Cancer Stem Cells; DAMPs: Damage-Associated Molecular Patterns; DMSO: Dimethyl Sulfoxide; E:T ratio: Effector-to-Target Ratio; FACS: Fluorescence-Activated Cell Sorting; Fas (CD95): Fas Cell Surface Death Receptor; FasL (CD95L): Fas Ligand; FBS: Fetal Bovine Serum; FITC: Fluorescein Isothiocyanate; GAPDH: Glyceraldehyde-3-Phosphate Dehydrogenase; γδT: Gamma Delta T Cells (γδT Cells); IC₅₀: Half-Maximal Inhibitory Concentration; IFN-γ: Interferon Gamma; IL: Interleukin; MCF-7: Michigan Cancer Foundation-7 Breast Cancer Cell Line; MDSCs: Myeloid-Derived Suppressor Cells; MET: Metformin; MHC: Major Histocompatibility Complex; mRNA: Messenger RNA; NK cells: Natural Killer Cells; PBS: Phosphate-Buffered Saline; PBMCs: Peripheral Blood Mononuclear Cells; PI: Propidium Iodide; qPCR/RT-qPCR: Quantitative Polymerase Chain Reaction/Reverse Transcription Quantitative PCR; RNA: Ribonucleic Acid; SD: Standard Deviation; TCR: T Cell Receptor; TME: Tumor Microenvironment; TNF-α: Tumor Necrosis Factor Alpha; Tregs: Regulatory T Cells
Acknowledgments
None.
Author’s contributions
N.C.T. conceived the study, performed or participated in all experiments, analyzed the data, and drafted the manuscript. N.T.H., N.T.D., and D.T.H.T. analyzed the transcription, translate of immune modulatory molecules. P.V.P. revised the manuscript and provided conceptual guidance. All authors read and approved the final manuscript.
Funding
This research is funded by Vietnam National University, Ho Chi Minh City (VNU-HCM) under grant number CB2025-18-26: “Evaluation of the effect of Paracetamol on the immune escape of breast cancer cells”, Project leader: MSc. Truong Chau Nhat within the framework of the Program titled "Strengthening the capacity for education and basic scientific research integrated with strategic technologies at VNU-HCM, aiming to achieve advanced standards comparable to regional and global levels during the 2025-2030 period, with a vision toward 2045. This research is also funded by VNUHCM-US Stem Cell Institute, University of Science, VNU-HCM, Viet Nam under grant number NCKH-SCI.09/25”.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Peripheral venous blood (15 mL) was collected from healthy adult donors. The collection protocol was strictly conducted in adherence to the Declaration of Helsinki and was approved by the Institutional Ethics Committee (NCKH-SCI.09/25). Written informed consent was obtained from all participants prior to inclusion.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the manuscript preparation process
During the preparation of this work the author(s) used Gemini in order to improve the language and readability of their paper. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
Competing interests
The authors declare that they have no competing interests.