


Docking-based virtual screening in search for natural PTP1B inhibitors in treating type-2 diabetes mellitus and obesity
- School of Pharmaceutical Sciences, Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia
- USM-RIKEN Centre for Aging Science (URICAS), Universiti Sains Malaysia, 11800 Minden, Penang, Malaysia
Abstract
Introduction: Growing incidence of type-2 diabetes mellitus (T2DM), together with obesity, shows the complexity and progressive nature of these metabolic disorders and alarms the necessity to explore new and alternative therapeutic pathways and drugs. Diabetes has also been proven to be a key cause of premature aging through different mechanisms. Understanding these mechanisms could lead us to manage diabetes more efficiently, thus curtailing its age-related complications. Insulin and leptin resistance are the most common pathophysiological link between T2DM and obesity. Protein tyrosine phosphatase 1B (PTP1B) is thought to interfere with glucose homeostasis and satiety through down-regulation of insulin and leptin signaling pathways. Thus, drugs that are potent to impede this enzyme should be effective in treating T2DM and obesity.
Method: In line with that, our current study involved screening of potent PTP1B inhibitors from the Natural Product Discovery System (NADI) database using in silico virtual screening and in vitro PTP1B inhibition study. The compounds that showed promising interaction with PTP1B catalytic site were traced down to their plant of origin. Further, the extracts of the plants were tested in vitro for PTP1B inhibition activity.
Results: Our results showed promising PTP1B inhibition activity of Pandanus amaryllifolius, Vitex negundo and Piper nigrum with 94.38%, 89.03% and 81.39% inhibition, respectively.
Conclusion: Therefore, the compounds of these plants might be an attractive option to develop drugs for T2DM and obesity.
Introduction
Type-2 Diabetes Mellitus (T2DM), together with obesity, is a growing threat to public health and accounts for multitudes of chronic deaths worldwide each year. According to the World Health Organization (WHO), the toll of diabetes incidence have grown exponentially from 108 million in the 1980s to 422 million in 20141 and is expected to exceed 600 million by 20352. Even children were affected with diabetes as early as preschool years4, 3. More than 90 percent of these diabetes cases were reported to be T2DM, which is the ultimate result of insulin resistance causing abnormal glucose homeostasis. Metabolic insulin signal transduction is initiated by the tyrosine phosphorylation of insulin receptor, which then recruits a series of downstream signaling molecules5, starting from insulin receptor substrate (IRS) to glucose transporter type 4 (GLUT4), which helps transport circulating glucose into cells in need of it. Signal arrest at any point of these signaling cascade events may lead to insulin resistance followed by an aggravation in blood glucose level. Leptin resistance, metabolic inflammation, mitochondrial disfunction and endoplasmic reticulum stress, which are the characteristics of obesity too, contribute to insulin resistance7, 6. Thus, insulin resistance has been regarded to be the most common pathophysiological link between T2DM and obesity.
Besides lifestyle modification, pharmacological interventions can be of great help in finding a prominent solution to this life-threatening metabolic disorder. Throughout these recent decades, many therapeutic approaches have become available as the traditional insulin therapy, on its own, have not been sufficient to enhance insulin sensitivity. Injectable insulin and oral anti-diabetic agents (OAD), such as metformin, sulfonylureas (or the structurally similar meglitinides), gliflozins, dipeptidyl peptidase-IV inhibitors, new insulin analogues (lispro, aspart and glargine), inhalational insulin drug, bromocriptine, thiazolidinediones (pioglitazone is the only one currently available), incretin mimetics and α-glucosidase inhibitors, are among the few current treatments12, 11, 10, 9, 8. There are several limitations, however, when using these drugs either in mono- or poly-therapy. The side effects, for instance, have been well-described14, 13, 11. Besides this, different responses to OAD among patients have also been recorded due to an array of single-nucleotide polymorphisms (SNPs) at several important genes such as insulin, leptin and sodium-glucose co-transporter-2 (SGLT2)8. Due to the complexity and progressive nature of T2DM, it is essential to explore new and alternative therapeutic pathways and drugs.
Protein tyrosine phosphatases (PTPs) have received immense attention since 1988 for their role in biological systems- as either positive or negative regulators of many important signal transductions15. Among the hundreds of phosphatases that exist, protein tyrosine phosphatase 1B (PTP1B) was the first enzyme to be purified and studied16. The involvement of this enzyme in many cellular signaling events, especially in the downregulation of insulin and leptin signaling pathways, have led to novel approaches for diabetes and obesity treatment17. In line with that, many research studies have been carried out in search for novel phosphotyrosine mimetics, such as lead PTP1B inhibitors. PTP1B inhibitors might potentially reverse insulin and leptin resistance and normalize plasma leptin, glucose and insulin18. Therefore, PTP1B could be a convincing candidate at the genetic, molecular, biochemical and physiological levels for drug design in combating the global epidemic of diabetes and obesity.
Natural products and traditional medicines serve a promising platform for drug discovery with a wide range of secondary metabolites, enabling access to various lead compounds. Many well-accepted drugs available on the markets today have been developed from natural products19. Taxol from Taxus brevifolia20, Orlistat, a derivative of lipstatin from bacterium Streptomyces toxytricini21, vincristine from Vinca rosea22, morphine from Papaver somniferum23, berberine from Berberis vulgaris24, and papaverine from opium poppy25 are a few examples of potent drugs that have emerged from natural products. Following that, our current study involved screening of potent PTP1B inhibitors from the Natural Product Discovery System (NADI) database (http://www.nadi-discovery.com) using in silico virtual screening. Plant species whose compounds give best hits and strong interactions were shortlisted for an in vitro assessment against PTP1B enzyme.
Methods
Hardware and software
The protein structure, 1C83, from PDB was used as the target protein for in silico screening using molecular docking. Hewlett-Packard PC with Core2 Duo processor 2.93 GHz, 2 GB of Random Access Memory (RAM) with operating system Linux Fedora Core 13 and Windows7 were used in this section of the project. ChemDraw Ultra 8.0 from ChemOffice 2004 was used to illustrate ligand structure, AutoDock Tools version 1.5.6 and AutoDock 4.226 were used in molecular docking simulation and ligand-based virtual screening, Discovery Studio 2.5 and Discovery Studio 4.0 Client from BIOVIA were used in the visualization of protein-ligand interactions.
Virtual screening
PTP1B enzyme with PDB ID 1C83 (1.80 Å) with ligand OAI (6-(oxalyl-amino)-1h-indole-5-carboxylic acid) was downloaded from protein databank (PDB; www.rcsb.org/pdb)27. The OAI was isolated from 1C83 crystal structure and saved as PDB file using Discovery studio 2.5 before being assigned with Gasteiger charges using AutoDockTools 1.5.6. The non-polar hydrogen atoms of the protein were merged and Gasteiger charges were assigned using the same program. A grid map (of 62 X 62 X 62 Å grid points at 0.375 Å spacing with center x= 44.746, y= 13.645, z= 2.842) was assigned to control docking to validate the parameter to be used for virtual screening, provided that the RMSD (root-mean-square deviation) value of the complex was to be less than 2.0 Å27. Then, virtual screening was performed against the NADI database using the control docking parameters. A total of 4000 natural compounds were screened and the compounds were shortlisted based on their favorable FEB (free energy of binding) and interactions with PTP1B. Fifty top-ranked compounds with FEB lower than -8.00 kcal/mol were shortlisted as potential hits and their interaction with PTP1B was cross-examined with control and literature. The hit compounds with preferable protein-ligand interaction were further clustered according to their plant of origin. Lastly, nine plant species (with different plant parts) were selected based on their availability and toxic properties. Due to the unavailability of Piper longum, we included Piper nigrum (which is the closest relative to P. longum and possesses the same phytochemical that showed the highest hit value in virtual screening, chabamide28) in the list of selected plants for the in vitro assessment. An addition of seven plant species with high fit values and good inhibition characteristics in pharmacophore study (results were not included in this current article) were also evaluated by in vitro studies.
Plant extracts
Methanol extracts of Momordica charantia (fruit), Oryza sativa (whole plant), Morinda citrifolia (leaf), Cymbopogon nardus (peel), Psidium guajava (leaf), Myristica fragrans (fruit), Murraya koenigii (leaf), Artocarpus heterophyllus (leaf), Piper nigrum (fruit), Artocarpus heterophyllus (bark), Anacardium occidentale (leaf), Barringtonia asiatica (leaf), Vitex negundo (leaf), Manilkara zapota (leaf) and Pandanus amaryllifolius (leaf), as well as the aqueous extracts of Manilkara zapota (fruit), Cymbopogon nardus (leaf), Calophyllum inophyllum (leaf) and Garcinia mangostana (leaf), were obtained from the Malaysian Institute of Pharmaceuticals & Nutraceuticals (IPHARM).
In vitro PTP1B inhibition assay
The PTP1B assay kit (Cat. No.: 539736-1KIT) was purchased from Merck (Kenilworth, NJ, USA). The test was carried out according to the manufacturer’s protocol. Briefly, 5 μL PTP1B enzyme at 0.5 ng/μL (final quantity was 2.5 ng per well) and 50 μL IR5 substrate solution at 150 μM (final concentration in each well was 75 μM) were prepared in assay buffer and added into the wells of a 96-well microtiter plate (provided in the kit). Instantly, 2.5 μL crude extracts dissolved in 2.5% DMSO (dimethylsulfoxide) at 4000 ppm were added to the wells with the reaction mixture to give a final concentration of 100 ppm. Assay buffer was added into each reaction mixture so that the total volume in the well was 100 μL. The mixture was incubated for 30 minutes at 30°C. Red Reagent was pipetted into each well to terminate the enzyme-substrate reaction after 30 min, and incubated for another 30 min at 30°C before the absorbance was measured. “Time zero” absorbance was measured by mixing 25 μL Red Reagent directly with 50 μL PTP1B enzyme at a concentration 1 ng/μL, with 50 μL IR5 substrate solution at 150 μM. Suramin and plain assay buffer were used as positive and negative controls, respectively, while plain 2.5% DMSO was used as blank or sample control. Absorbances were converted to nmol POby using the standard curve of phosphate. Lastly, the percentage of inhibition was calculated using the equation below:
The interactions between PBD structure 1C83 and OAI
| Type of interaction | Distance (Å) | Interacting Residues(H-Donor to H-Acceptor) | OAI’s atom | Amino Acid Residue |
|---|---|---|---|---|
| Hydrogen Bond | 2.70 | A:LYS120:NZ –A:OAI301:O20 | O20 | NZ:LYS120 |
| Hydrogen Bond | 2.88 | A:SER216:N –A:OAI301:O16 | O16 | N:SER216 |
| Hydrogen Bond | 3.33 | A:ALA217:N –A:OAI301:O16 | O16 | N:ALA217 |
| Hydrogen Bond | 2.75 | A:GLY220:N –A:OAI301:O17 | O17 | N:GLY220 |
| Hydrogen Bond | 3.07 | A:ARG221:N –A:OAI301:O15 | O15 | N:ARG221 |
| Hydrogen Bond | 2.78 | A:ARG221:NE - A:OAI301:O15 | O15 | NE:ARG221 |
| Hydrogen Bond | 3.14 | A:ARG221:NH2 - A:OAI301:O16 | O16 | NH2:ARG221 |
| Hydrogen Bond | 3.24 | A:SER216:CB –A:OAI301:O19 | O19 | CB:SER216 |
| Hydrophobic | 4.80 | A:OAI301 –A:VAL49 | Pi-Orbital | Alkyl:VAL49 |
| Hydrophobic | 4.74 | A:OAI301 –A:ALA217 | Pi-Orbital | Alkyl:ALA217 |

OAI (stick presentation) interacting with PTP1B (ribbon presentation) residues. Interacting PTP1B regions were coloured in yellow.
RESULTS
Control docking and virtual screening
From a total of 126 registered PTP1B crystal structures, the structures with PDBID: 1C83 and 1C85 were shortlisted after comparing the X-ray resolution, classification of enzyme, and the objectives of the original literature. These structures were studied with the presence of ligands that inhibited PTP1B. Despite the comparably low R-value and RFree-value of 1C83 and 1C85 ranging from 0.183 to 0.197 and 0.231 to 0.267, respectively, the high-resolution structure 1C83 (1.80 Å) was favored, as compared to 1C85 (2.75 Å). The interactions between protein crystal 1C83 with OAI (Figure 1 and 2) are listed in Table 1. Control docking was performed to confirm the accuracy of parameters to be used in docking simulation (Figure 1). A dock conformation (Run 21) in the top ranked cluster with the lowest binding energy of -10.15 kcal/mol, mean binding energy of -9.45 kcal/mol, and reference RMSD of 0.50 Å was obtained. The low reference RMSD (<2.0 Å) indicated only a minute difference29 between the docked pose and originally published pose30. Therefore, the docking parameters can mimic the experimental condition to reproduce a similar docking environment and were used for further virtual screening. Figure 2 showed the stacking of OAIs in the original protein crystal pose (yellow) and re-docked pose (green).

The position of OAIs in original protein crystal and control docking. Yellow OAI represented original pose in protein crystal and green OAI represented re-docked pose.
Fifty top ranked ligands with their originating plants in virtual screening study against 1C83
| NADI Code | Lowest Binding Energy | Scientific name | Compound name |
|---|---|---|---|
| MSC759 | -10.03 | Piper longum | Chabamide |
| MSC3271 | -9.87 | Dryobalanops aromatica | α-Viniferin |
| MSC2289 | -9.68 | Momordica charantia | Kuguacin B |
| MSC665 | -9.67 | Stemona tuberosa | 9α-bisdehydrotuberostemonine A |
| MSC341 | -9.56 | Garcinia mangostana | Garcinone E |
| MSC3480 | -9.53 | Cymbopogon nardus | 6C-pentosyl luteolin |
| MSC2437 | -9.53 | Calophyllum inophyllum | Caloxanthone B |
| MSC3270 | -9.43 | Shorea gibbosa | Ampelopsin A |
| MSC770 | -9.32 | Clausena excavata Burm. | Clausarin |
| MSC3221 | -9.25 | Jatropha Curcas | 24 -Methylenecycloartenol |
| MSC254 | -9.20 | Garcinia mangostana | Mangostenone B |
| MSC1647 | -9.14 | Manilkara zapota | 23,26-Dihydroxy-tirucalla-7,24-dien-3-one |
| MSC3019 | -9.14 | Dysoxylum hainanense | α-Spinasterol acetate |
| MSC1383 | -9.13 | Vitex trifolia | 17-6-Acetoxy-9-hydroxy-13(14)-labden-16,15-olide |
| MSC606 | -9.11 | Oryza sativa | n-β-D-Glucopyranosyl-glutamate-indole-3-acetic acid |
| MSC348 | -9.09 | Garcinia mangostana | 1,3,7-Trihydroxy-2,8-di-(3-methylbut-2-enyl)xanthone |
| MSC1946 | -9.07 | Brucea javanica | Dihydrobruceajavanin A |
| MSC248 | -9.06 | Morinda citrifolia | Caloxanthone A |
| MSC1670 | -9.06 | Calophyllum inophyllum | Americanin A |
| MSC347 | -9.03 | Garcinia mangostana | Tovophyllin B |
| MSC607 | -8.91 | Oryza sativa | n-β-D-Glucopyranosyl-aspartate-indole-3-acetic acid |
| MSC2036 | -8.90 | Tinospora tuberculata | Cycloeucalenone |
| MSC2297 | -8.87 | Momordica charantia | Kuguacin H |
| MSC2101 | -8.87 | Boesenbergia rotunda | Rotundol |
| MSC645 | -8.86 | Stemona tuberosa | Bisdehydrostemoninines A |
| MSC664 | -8.85 | Stemona tuberosa | 9α-Bisdehydrotuberostemonine |
| MSC767 | -8.85 | Caesalpinia pulcherrima | 6β-Cinnamoyl-7β-hydroxyvouacapen-5α-ol |
| MSC2308 | -8.82 | Momordica charantia | Kuguacin S |
| MSC2081 | -8.81 | Boesenbergia rotunda | Nicolaioidesin C |
| MSC611 | -8.77 | Oryza sativa | Ergosterol peroxide |
| MSC1238 | -8.76 | Blumea balsamifera | 3-O-7''-biluteolin |
| MSC1047 | -8.75 | Jasminum sambac | Sambacin |
| MSC3497 | -8.74 | Calophyllum inophyllum | Caloxanthone O |
| MSC2067 | -8.73 | Boesenbergia rotunda | Panduratin B1 |
| MSC2656 | -8.72 | Murraya paniculata | Genistin-6'-O-malonate |
| MSC1665 | -8.71 | Eugenia cumini | 2-Hydroxy-3-(hydroxymethyl)anthraquinone |
| MSC1254 | -8.71 | Morinda citrifolia | Myricetin 3-O-(4''-O-acetyl)-α-L-rhamnopyranoside |
| MSC3241 | -8.69 | Tinospora tuberculata | Diptoindonesin G |
| MSC2748 | -8.69 | Etlingera elatior | 16-Hydroxylabda-8(17),11,13-trien-16,15-olide |
| MSC2035 | -8.69 | Hopea mengarawan | Cycloeucalenol |
| MSC3461 | -8.67 | Baeckea frutescens | Inophyllin A |
| MSC3418 | -8.67 | Calophyllum inophyllum | 6-Methylquercetin |
| MSC735 | -8.65 | Piper sarmentosum | Horsfieldin |
| MSC1639 | -8.65 | Dysoxylum hainanense | ent-18-Acetoxy-16-hydroxy-8(14)-pimaren-15-one |
| MSC2567 | -8.62 | Murraya paniculata | Marmesin-4'-O-α-L-arabinopyranoside |
| MSC876 | -8.61 | Pandanus amaryllifolius | Pandamarilactonine-B |
| MSC1369 | -8.60 | Boesenbergia rotunda | Cudraflavone C |
| MSC2068 | -8.60 | Artocarpus champeden | Panduratin B2 |
| MSC659 | -8.59 | Stemona tuberosa | Tuberocrooline |
| MSC2296 | -8.59 | Momordica charantia | Kuguacin G |
Inhibitory activity of selected plant extracts using 2.5% DMSO as solvent against PTP1B
| Sample | % Inhibition | Sample | % Inhibition |
|---|---|---|---|
| Suramin | 87.54 ± 1.02 | Manilkara zapota (L) | 39.16 ± 18.88 |
| P. amaryllifolius (L) | 94.38 ± 2.43 | Psidium guajava (L) | 30.73 ± 8.22 |
| Vitex negundo (L) | 89.03 ± 3.65 | Oryza sativa (WP) | 30.24 ± 6.75 |
| Piper nigrum (F) | 81.39 ± 10.38 | Garcinia mangostana (L) | 25.54 ± 15.98 |
| Cymbopogon nardus (L) | 79.78 ± 6.12 | Artocarpus heterophyllus (B) | 9.83 ± 32.23 |
| Cymbopogon nardus (P) | 66.90 ± 6.43 | Myristica fragrans (F) | 2.81 ± 6.97 |
| Cymbopogon nardus (L) | 66.02 ± 13.20 | Barringtonia asiatica (F) | 2.28 ± 23.82 |
| Manilkara zapota (F) | 62.06 ± 24.87 | Murraya koenigii (L) | CBD |
| Calophyllum inophyllum (L) | 48.58 ± 21.05 | Artocarpus heterophyllus (L) | CBD |
| Momordica charantia (F) | 42.80 ± 14.62 | Anacardium occidentale (L) | CBD |
| Morinda citrifolia (L) | 42.18 ± 7.71 | - | - |

Docking conformations of five top ranked ligands with important PTP1B residues. The order of ranking of compounds from highest to lowest: MSC759 (green) > MSC2289 (red) >MSC665 (purple) > 341 (blue) > MSC2437 (dark green).
The validated docking parameters were used to screen 4000 natural compounds from NADI database through virtual screening. The highest ranked ligand was MSC759 with FEB of -10.03 kcal/mol, comparable to the binding of OAI (-10.15 kcal/mol). From a pool of 148 ligands (4.93% from sample pool) that exhibited FEB higher than -8.00 kcal/mol (±2 kcal/mol from control)31, 26, we grouped 50 ligands with top hits and documented them in Table 2, together with their respective plants of origin as listed in NADI. All these 50 ligands were docked inside the catalytic site of PTP1B in order to scrutinize the protein-ligand interactions and the data were cross-examined and compared to the control and literature. Figure 3 showed that the docking conformations of the 5 top ranked ligands inside the binding site were similar to the docking conformation of the control, even though their FEB varied. The compounds that were listed among the 50 top hits and which showed preferable protein-ligand interactions are illustrated in Figure 4. Their interactions with PTP1B are detailed in Figure 5 and Figure 6.

Ligands with best interaction with PTP1B in molecular docking virtual screening study.

The interactions of top ranked ligands including MSC759 (Ranked 1st), MSC2289 (Ranked 3rd), MSC341 (Ranked 5th) and MSC2437 (Ranked 6th). The coloured dotted lines representing interactions between ligand and residue. Green line represented hydrogen bonding, blue line represented unconventional carbonyl hydrogen bonding/interaction, orange line represented electrostatic attraction, red line represented Pi-Pi stacking and purple line represented Pi-Alkyl interaction.
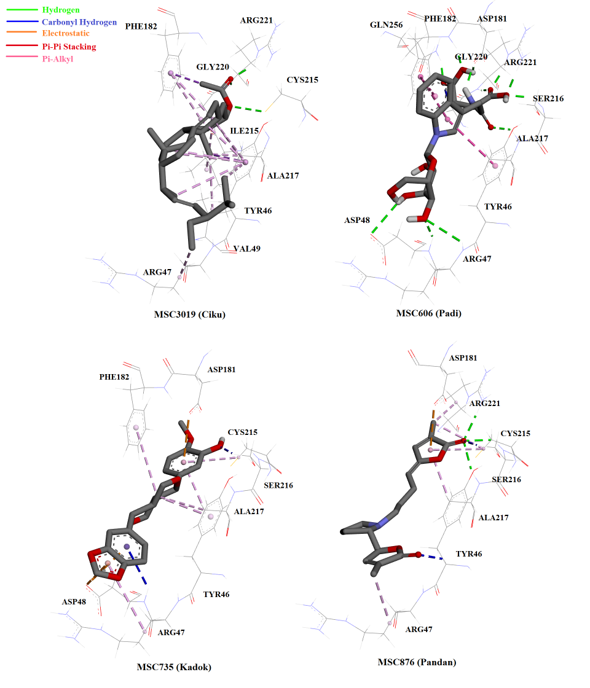
The interactions of top ranked ligands including MSC3019 (Ranked 12th), MSC606 (Ranked 15th), MSC735 (Ranked 43rd) and MSC876 (Ranked 46th). The colored dotted lines representing interactions between ligand and residue. Green line represented hydrogen bonding, blue line represented unconventional carbonyl hydrogen bonding/interaction, orange line represented electrostatic attraction, red line represented Pi-Pi stacking and purple line represented Pi-Alkyl interaction.
In vitro PTP1B inhibition assay
The selected 20 crude extracts were screened for their in vitro PTP1B inhibition activity at 100 ppm. The results are listed in Table 3. As can be seen, 7 crude extracts showed inhibition activity above 50%, with the highest inhibition recorded for the methanol leaf extract of P. amaryllifolius followed by V. negundo and P. nigrum, which exerted 94.38, 89.03% and 81.39% inhibition, respectively. These inhibition activities were almost as strong as the PTP1B-inhibitor included in the assay kit, suramin, which gave an inhibition of 87.54%. Among the extracts tested, methanol leaf extracts of M. koeniggi, A. heterophyllus and A. occidentale showed the lowest or non-detectable inhibition activity against PTP1B enzyme. On the other hand, extracts of A. heterophyllus, M. fragrance and B. asiatica showed inhibition activity less than 10%, which indicated poor PTP1B inhibition strength. A moderate inhibition activity ranging from 42% to 48% was observed with C. inophyllum (L), M. charantia (F) and M. citrifolia (L) extracts.
DISCUSSION
Screening for effective drugs can be staggeringly time-consuming and costly. In fact, the success rate that the tested molecule turns out to have activity at the biological target of interest lies below 0.5%32. The author described the process of screening for a drug likened to searching for a needle in a haystack. Computational aided methods of drug design has become a promising approach to research since late 1990’s33. Since then, advancements in cheminformatics and Computer-Aided Drug Designing (CADD) have revolutionized the process of drug discovery into a fast, cost-effective and reliable approach. Such approaches are reasonably more efficient such that they can be used as a scoring function to select the small molecules, among millions or even billions of secondary molecules thought to exist, with high affinity towards biological targets35, 34. Furthermore, through computer modeling, the small molecules or ligands can be optimized, tested for side effects, or even modified to yield properties more akin to the ideal drug. The success story of CADD in many research discoveries have been reviewed38, 37, 36. Herein, we applied virtual screening through molecular docking in search for promising PTP1B phosphatase inhibitors among 4000 natural compounds from the NADI database, which we believe could serve as a preliminary step for future drug development for T2DM and obesity.
From the 4000 natural compounds screened, 50 compounds with the highest hits were redocked back into PTP1B crystal structure, 1C83, to inspect the interaction between PTP1B and each compound. The catalytic domain of PTP1B enzyme is comprised of the amino acids 30-278. From these, residue Cys215 (active site nucleophile), WPD loop (amino acids 79-187), YRD loop (Tyr46 and charged residues: Arg47 and Asp48), and the secondary aryl-phosphate binding site were reported to be the most significant regions in the PTP1B enzyme structure39. Residues His214-Arg221 (His-Cys-Ser-Ala-Gly-Ile-Gly-Arg) that surrounded the catalytic residue Cys215 form a cradle-like structure that favors substrate binding40. Meanwhile, WPD loop acts like an arm that can be either found in closed or opened motif with regards to substrate binding to the enzyme. In addition to assisting in stronger binding of substrate to enzyme, the WPD loop can be involved in catalysis by protonating the tyrosyl leaving group of the substrate by Asp18141. On the contrary, the charged residues in the YRD loop is known to be important in inhibitor specificity (via a salt bridge formation between the residues) to potent inhibitors that have basic nitrogen, thus offering selective inhibition over other PTPs43, 42.
Most of the compounds from the NADI database bound to the PTP1B catalytic site (similarly to OAI) and interacted with amino acids at the catalytic site and surrounding active site loops, WPD and YRD (Figure 4). Chabamide (MSC759; Piper longum), the compound with the highest hit in the virtual screening interacted with three amino acids at the catalytic region of PTP1B through hydrogen bonding: Arg47, Arg221, and Gly221 (Figure 5). Conformation with the lowest FEB (i.e. highest affinity towards the enzyme) of this compound might be due to the interaction with Arg47, the charged residue in the YRD loop that is specific for inhibitor molecules. Furthermore, 6-C-pentosyl luteolin (MSC2437) from Cymbopogon citrates/nardus displayed four promising hydrogen bonding with Arg 47, Asp 181, Ser 216 and Arg 221. The compound that formed the greatest hydrogen bonding with the enzyme compared to OAI (inhibitor in the crystal structure) was n-β-D-Glucopyranosyl-glutamate-indole-3-acetic acid (MSC606), which was isolated from Oryza sativa. This ligand formed eight hydrogen interactions (with residues in the catalytic, cradle-like domain and in the charged region of YRD loop), thereby indicating the strong bonding with PTP1B (Figure 6).
Another compound that showed significant interaction with the enzyme was pandamarilactonine-B (MSC876), an alkaloid with two lactones and one pyrrolidine ring structure44, from Pandanus amaryllifolius. This ligand was the only one (among the 50 top hits) to form hydrogen bonding with Cys215, the catalytic residue (Figure 6). Upon binding, pandamarilactonine-B was clamped between Asp181 and Ala217 through pi-interaction, which showed good penetration into the catalytic site and inhibition characteristics through interference with protonation activity to form the cysteinyl phosphate intermediate41. In addition, a carbon-hydrogen bonding with Tyr46 and Arg47, the charged residues in YRD loop, was also observed between pandamarilactonine-B and PTP1B. Furthermore, kuguacin B (MSC2289) from Momordica charantia, a-spinasterol acetate (MSC3019) from Manilkara zapota, horsfieldin (MSC735) from Cymbopogon nardus, and garcinone E (MSC341) from Garcinia mangostana all interacted with PTP1B through non-covalent bonding, such as pi-pi stacking and pi-alkyl interactions. These interactions were believed to be insignificant as they were considerably further compared to hydrogen bonding and electrostatic interactions45.
Taking into consideration the strength of protein-ligand interactions, we selected a few plants to be studied for in vitro PTP1B inhibition activity. Based on the test, methanol leaf extracts of P. amaryllifolius exerted the highest enzyme inhibition, which was never reported elsewhere, although P. amaryllifolius is known to be rich with nitrogen-containing alkaloids. As discussed earlier in this section, the nitrogen molecule could serve as the basic entity to interact with the charged YRD loop through a salt bridge formation43. In support of our observation, Sasidharan et al.46 have reported robust anti-hyperglycemic activity of ethanol extract of P. amaryllifolius in streptozotocin (STZ)-induced diabetic rats. Similar to P. amaryllifolius, V. negundo and P. nigrum showed greater than 80% inhibition. The anti-diabetic properties of V. negundo and P. nigrum have been well-documented by many research studies51, 50, 49, 48, 47, but most of them involved in vivo studies and no inhibitory pathways were illustrated. Betulinic acid, one of the compounds present in V. negundo leaves, was reported to have potent PTP1B inhibition activity52. Nonetheless, the methanol leaves and peel extracts of C. nardus, and aqueous leaves and fruit extracts of C. nardus and M. zapota, respectively, showed moderate activity towards PTP1B enzyme.
The computer-aided drug designing approach in the search of PTP1B inhibitors often targets the catalytic site of the protein. Though the catalytic region is important for the activity of the enzyme, looking outside of the catalytic pocket of the protein structure may expedite the discovery of drugs with more selectivity towards PTP1B. This is important since all PTPs share nearly 50-80% homology in the catalytic domain which could be a challenge in providing selectivity to PTP1B53. Moreover, the charged catalytic domain (YRD loop) poses a challenge in designing PTP1B inhibitors, such as oral drugs. It is for these reasons that many computationally designed and validated drugs have failed to yield the same expected outcomes in vivo. The non-catalytic phosphotyrosine-binding domain offers an interesting and alternative option to target PTP1B54 and should be considered for drug development.
CONCLUSION
Our virtual screening study and enzymatic assays indicated the promising PTP1B inhibitory activity of Pandanus amaryllifolius leaves, Vitex negundo leaves and Piper nigrum fruit, Cymbopogon nardus (aqueous and methanol extracts of leaves and peel), and Manilkara zapota leaves. The good correlation between our in silico method and enzyme assay shows the validity of our test parameters. Moreover, cross-examination and comparison of our results to the published literature show complementary ethnopharmaceutical properties of the genus Pandanus, Piper and Cymbopogon. Further fractionation or isolation of active principles from these plants can provide a good platform to develop promising anti-diabetic or anti-obesity drugs through PTP1B-targeted approaches. However, taken into account the limitations of targeting only the catalytic region of PTP1B, future investigations should include all possible binding pockets.
Abbreviations
CADD: Computer-aided drug designing
DMSO: Dimethyl sulfoxide
FEB: Free energy of binding
GLUT4: Glucose transporter type 4
IPHARM: Institute of Pharmaceuticals & Nutraceuticals
IRS: Insulin receptor substrate
NADI: Natural product discovery system
OAD: Oral anti-diabetic agents
PTP1B: Protein tyrosine phosphatase 1B
PTPs: Protein tyrosine phosphatases
RAM: Random access memory
RMSD: Root-mean-square deviation
SGLT2: Sodium-glucose co-transporter-2
SNP: Single-nucleotide polymorphism
STZ: Streptozotocin
T2DM: Type-2 diabetes mellitus
WHO: World Health Organization
Competing Interests
Authors declare that there are no conflicts of interests.
Authors' Contributions
Ho Yueng Hsing carried out the experiment. Selestin Rathnasamy wrote the manuscript with the assistant of Roza Dianita. Habibah A. Wahab edited the manuscript and supervised the project.
Acknowledgment
We thank to USM and RIKEN for financial support from USM RU TOP-DOWN project entitled Catalogue of USM-RIKEN Natural Product (CURINaP) Library for the Discovery of Bioactive Molecules on Ageing and Ageing-Related Diseases, 1001/PFARMASI/870031.