

Dysregulated Metabolism in Cancer
- Advanced Medical and Dental Institute, Universiti Sains Malaysia 13200 Bertam Kepala Batas, Penang Malaysia
Abstract
Metabolism describes the cellular bioenergetic pathways that provide energy and macromolecules for protein, lipid, and nucleic acid syntheses. In cancer, malignant cells alter the metabolic pathways to acquire nutrients needed for proliferation and survival. The various metabolic modifications exhibited by cancer cells include aerobic glycolysis, decreased oxidative phosphorylation, and elevated production of biosynthetic intermediates. To support the increasing need for these metabolic modifications, cancer cells increase the expressions of plasma membrane transporters and enzymes involved in the metabolic pathways. Additionally, some cancer cells escape chemotherapy treatment by reprogramming their metabolic activities. This chemotherapy-induced resistance mechanism allows malignant cells to promote their survival as well as to provide defense against cell damage by engaging various metabolic shunt pathways. Therefore, understanding metabolic reprogramming in cancer may provide useful information that can further be exploited to strategize potential treatment interventions and subsequently foster better outcomes among patients with cancer.
Introduction
Some of the characteristics that distinguish cancer cells from normal cells are high proliferation activity, self-renewal property, and cell death evasion1, 2. As cells evolve toward neoplastic characteristics, they modify nutrient acquisition mechanisms as well as rewire bioenergetic pathways and undergo metabolic changes.
Dysregulated metabolism is one of the seven hallmarks of cancer and is rooted back to the study by Otto Warburg in 19561, 2, 3. In the study, he observed an abnormal metabolic phenomenon exhibited by cancer cells, in which tumors utilized glycolysis to produce energy in the presence of oxygen1. This observation opposes the normal energy production in normal cells whereby the cells prefer to employ oxidative phosphorylation (OXPHOS) over glycolysis when oxygen is available. Since then, research on cancer metabolism has been growing steadily with less emphasis given on the area. However, in the last decade, the field has emerged as an area of interest among scientists and clinicians to unravel the link between dysregulated metabolic activity and cancer2. For instance, studies have found that during transformation, cancer cells require continuous bioenergetic reactions and copious amounts of ATPs to guarantee a sufficient supply of energy4, 5, 6. Accordingly, cancer cells adopt numerous metabolic strategies to support the need for energy, such as higher uptake of nutrients, execution of different shunt pathways, and overexpression of several metabolic enzymes and proteins7, 8, 9, 10.
More recently, the roles of dysregulated metabolism in cancer have also been speculated to underlie the disease resistance to therapy. Emerging evidence has pointed toward metabolic reprogramming in resistant cancer cells when they are exposed to selective pharmacological or molecular inhibitions11, 12, 13. For example, a portion of surviving cancer cells treated with a standard chemotherapy agent shunted their metabolic activities toward building of membrane skeletons and nucleotide blocks and remained dormant for a certain period12, 14, 15. This phase of dormancy prepares the cells to emerge as resistant clones and cause relapse in the future14. Therefore, this review aimed to discuss the fundamental role of metabolism in cancer and present recent findings on the rewiring of metabolic activity when tumor cells evolve toward resistancy. Targeting metabolism by inhibiting known metabolic pathways and issues surrounding this approach are also outlined in this review.
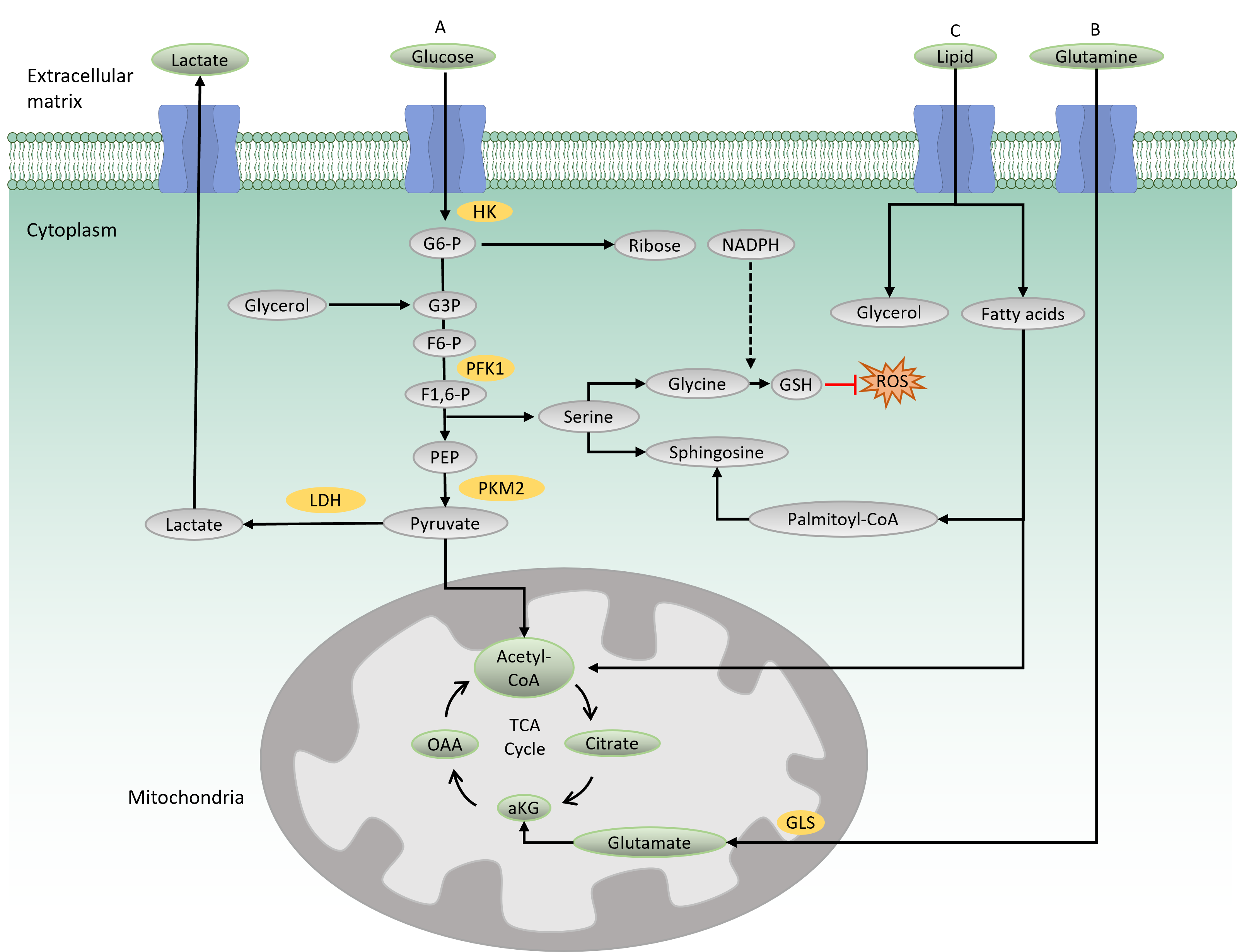
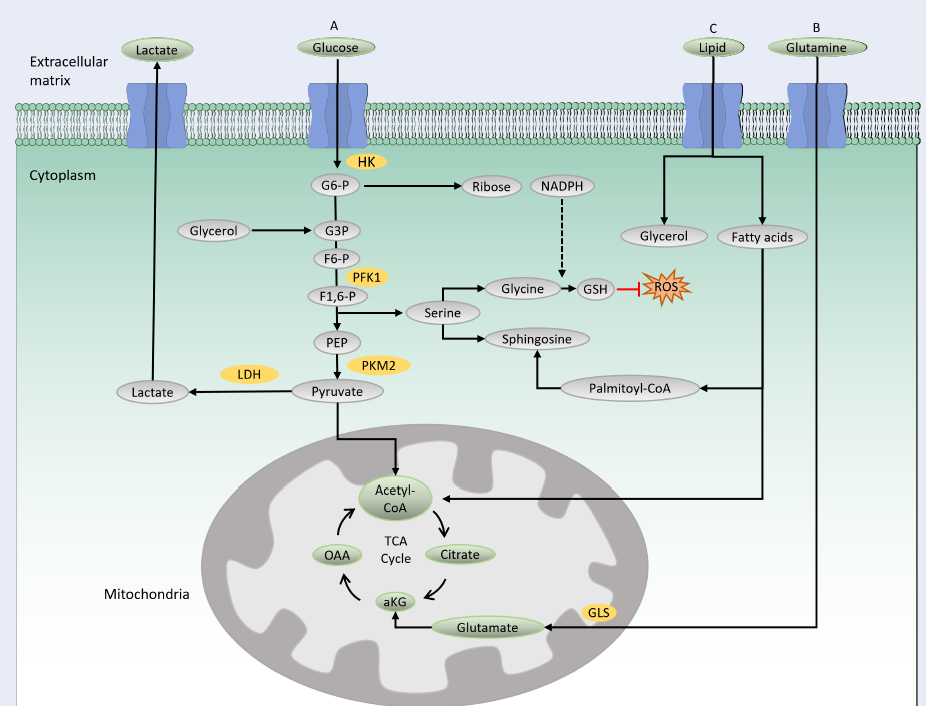
Overview of the metabolic pathways. The substrates for metabolism include glucose, glutamine and lipid. A) Glucose is the substrate for glycolysis and is transported into the cells through glucose transporter. It is phosphorylated by HK to produce G6P that prevents diffusion of the molecule to the outer membrane. G6P can enter pentose phosphate pathway to produce backbone for nucleotide synthesis and reducing agent, NADPH. PFK1 catalyses the conversion of F6P to F1,6P. Glycolysis intermediate can also execute SSP. Serine is the precursor for the synthesis of GSH as well as sphingiosine. GSH protects cells from oxidative damage by ROS. The final product of glycolysis is pyruvate that enters mitochondria for TCA cycle and complete oxidation for energy generation. Pyruvate can also be converted into lactate by LDH which later is transported to the extracellular matrix. B) Glutamine is transported into the cells by glutamine transporter and is converted into glutamate by GLS in the mitochondria. Glutamate is transformed into aKG which feeds into the TCA cycle. C) Lipid is broken down into fatty acids and glycerol in the cytoplasm. In mitochondria, beta-oxidation of fatty acids produces acetyl-CoA which feeds the TCA cycle. Fatty acids can also be transformed into palmitoyl-CoA that provides backbone for sphingosine. Sphingosine is used for the synthesis of membrane lipids. HK: hexokinase; G6P: glucose 6-phosphate; PFK1: phosphofructokinase; F6P: fructose 6-posphate; F1,6P: fructose 1,6-biphosphate; SSP: serine synthesis pathway; GSH: glutathione; ROS: reactive oxygen species; TCA: tricarboxylic cycle; LDH: lactate dehydrogenase; GLS: glutaminase; aKG: alpha-ketoglutarate.
Glycolysis
Glycolysis is the main catabolic pathway that breaks down glucose molecules into pyruvate following a series of enzymatic reactions. A complete glycolysis reaction processes 1 mole of glucose into 2 moles of ATP and 2 moles of NADH. In the first step of glycolysis, glucose is transported into the cytoplasm by a glucose transporter (GLUT) and is then phosphorylated by hexokinase, producing a negatively charged glucose 6-phosphate (Figure 1A). The negative charge prevents diffusion of the molecule through the cell membrane, thus trapping the molecule within the cytoplasm. Along the glycolysis pathway, phosphofructokinase 1 (PFK1), a rate-limiting enzyme, catalyzes the conversion of fructose 6-phosphate to fructose-1,6-biphosphate. In this process, ATP is consumed, making this reaction a crucial and irreversible step in the glycolysis pathway. The final product of glycolysis is pyruvate, a three-carbon molecule that serves as a carbon precursor for subsequent bioenergetic processes. Depending on the physiological condition, pyruvate has two fates: either to enter the mitochondria for the tricarboxylic acid (TCA) cycle and subsequent OXPHOS reaction or to be converted into lactate by lactate dehydrogenase16.
When the oxygen is less favorable, cells opt to convert pyruvate into lactate, which is then transported out to the extracellular matrix by a lactate transporter16. This process is termed aerobic glycolysis, which generates 2 ATPs per glucose molecule.
TCA and OXPHOS
In the presence of oxygen, pyruvate enters the mitochondria for complete catabolic and anabolic reactions in the TCA cycle. In the mitochondrial matrix, pyruvate is oxidized by coenzyme A (CoA) to produce acetyl-CoA. Acetyl-CoA then reacts with oxaloacetate to produce citrate, which later undergoes eight enzymatic reactions in the TCA cycle. The final product of the TCA cycle is oxaloacetate, which consequently feeds back into the cycle (Figure 1A). A complete TCA cycle produces a number of TCA cycle intermediates that serve as precursors for biosynthetic processes. The TCA cycle also produces reducing agents, NADH and FADH2, which are needed for electron transfer in the mitochondrial respiratory chain. In the presence of oxygen, this electron transport chain funnels electrons through the four complexes (I to IV) in the inner mitochondrial membrane to generate 32–34 ATPs per glucose molecule, making it an efficient energy production process (i.e., OXPHOS)17.
The TCA cycle is also fueled by glutamine-derived metabolites (Figure 1B). Glutamine provides both carbon and nitrogen precursors for cellular bioenergetics and biosynthesis. It is transported into the cells by glutamine transporters and transformed into glutamate by glutaminase (GLS), which is later converted to alpha-ketoglutarate and feeds back into the TCA cycle. Glutamine metabolism has been shown to maintain TCA cycle activity in cells with impaired mitochondria, thus providing crucial support for cell survival18.
Pentose Phosphate Pathway (PPP) and Serine Synthesis Pathway (SSP)
The PPP and SSP are two metabolic pathways parallel to glycolysis (Figure 1 A). The PPP (also known as the phosphogluconate or hexose pathway) produces the sugar–phosphate backbone for nucleic acid synthesis and NADPH for fatty acid synthesis and maintenance of reduction–oxidation under stress conditions 19. Meanwhile, the SSP produces serine, which is the source for biosynthesis of nonessential amino acids, glycine and cysteine. Glycine acts as a precursor for the production of glutathione (GSH), an antioxidant that provides defense from oxidative stress damage20. Serine provides a backbone for sphingosine in the synthesis of membrane lipids. Notably, PPP and SSP dysregulations have been reported to be involved in several types of cancer, such as breast and blood cancers8, 21.
Lipid Metabolism
Lipid metabolism is the process of synthesizing, storing, and degrading lipids (triglycerides, cholesterol, fatty acids, and phospholipids) to produce energy or structural components of cell membranes. In lipid catabolism, triglycerides are broken down into fatty acids and glycerol (Figure 1C). In the cytosol, glycerol is transformed to glyceraldehyde 3-phosphate, an intermediate in the glycolysis pathway that can further be oxidized to generate energy. In the mitochondria, beta-oxidation of fatty acid produces acetyl-CoA that feeds into the TCA cycle. Membrane lipid biosynthesis involves the synthesis of the membrane backbones, sphingosine or glycerol, followed by the addition of fatty acids to generate phosphatidic acid. Phosphatidic acid is modified by the addition of hydrophilic head groups to the membrane skeleton, making a complete structure of membrane lipids. Recently, lipid reprogramming has been shown to be responsible for the resistance mechanism seen in blood and solid tumors that have been treated with chemotherapeutic agents14, 22.
DYSREGULATED METABOLISM IN CANCER
In cancer, the transformed cells undergo a complex metabolic reprogramming activity to promote their increased need for energy. This alteration is manifested by an increased uptake of nutrients, such as glucose and glutamine, a high rate of glycolysis resulting in increased extracellular acidification, and a higher expression of metabolic enzymes and proteins5, 15, 23, 24.
An increased uptake of glucose is one of the characteristics of dysregulated metabolism in cancer7, 9, 23. In a study of breast cancer cells, upregulated glucose intake and increased production of both pyruvate and lactate were observed in an aggressive tumor model23. Furthermore, inhibition of the glycolytic function with glycolytic inhibitor 2-deoxyglucose diminished cell growth, whereas inhibition of the mitochondrial function with 3-nitropropionic acid, an inhibitor of the TCA cycle, did not reduce the invasiveness; these findings indicate the reliance of invasive characteristics on glycolysis but not on the mitochondrial energetic pathway23.
A high intake of glucose is supported by an increased expression of GLUTs and is linked to the oncogenic role of the transcription factor, NF-KB25, 26, 27. Among the 14 homologous families of GLUT proteins, GLUT1 and GLUT3 have been predominantly reported to be implicated in many types of cancer, such as breast cancer, colorectal carcinoma, leukemia, and glioblastoma7, 9, 26, 27, 28. High expressions of GLUT1 and GLUT3 are also associated with poor clinical outcomes in patients with colorectal carcinoma and glioblastoma7, 9.
The rate of glycolysis is increased in many types of cancer owing to overexpression of PFK129, 30, 31, 32. PFK1 has three isoforms: PFK1 platelet (PFKP), PFK1 muscle, and PFK1 liver, which are differentially expressed in different organs and tissues33, 34. However, PFKP is predominantly dysregulated in many types of cancer, including lung, breast, liver, and kidney cancers21, 30, 31, 32, 35. In a PTEN loss brain tumor model, AKT phosphorylated and stabilized PFKP and subsequently promoted glycolysis and tumor progression36. Furthermore, PFKP was reported to be involved in epithelial–mesenchymal transition in breast cancer cells, and its overexpression is a poor prognostic factor for patients with breast cancer with BRCA1 deficiency21, 37.
PKM2 is another glycolytic enzyme that functions at the end of the glycolytic pathway and is overexpressed in many types of cancer that exhibit a high glycolytic activity10, 38. It converts phosphoenolpyruvate to produce pyruvate and ATP, which gives advantage to tumor cells that have a higher expression of this enzyme, such as pancreatic cancer cells10. Intriguingly, PKM2 is translocated to the plasma membrane and provides exclusive ATP supply for ATP-dependent calcium pump, thus preventing cytoplasmic calcium overload and subsequent cell death10. Dysregulated PKM2 is also reported in cells with a compromised mitochondrial function, which hinders the glycolytic flux from entering the TCA cycle39.
Consequently, a high rate of glycolysis results in the accumulation of extracellular lactate, making the tumor microenvironment acidic and optimal for metastasis, vascularization, and resistance40. For example, an acidic microenvironment poses a growth advantage for aggressive colon and breast tumors in both in vitro and in vivo studies17, 40. A low-pH environment is also associated with drug resistance and poor drug efficacy as exhibited by acid-adapted colon carcinoma cells13. Correspondingly, cancer cells require adaptation to maintain a low extracellular pH. Thus, cancer cells increase the expression of various membrane transporters, including sodium–hydrogen exchanger-1 and monocarboxylate transporters (MCTs), which are directly linked to tumors with high metabolic rates, such as cervix squamous cell carcinoma and B-cell lymphoma40, 41, 42.
Cancer cells also rely on the PPP and SSP as alternatives for glycolysis and OXPHOS. For example, T-cell acute lymphoblastic leukemia cells suppress glycolysis and shift the glycolysis intermediates toward the PPP and SSP, both of which are important for providing skeletons for tumor growth as well as for protecting against oxidative damage8. Interestingly, glycolysis is suppressed owing to the phosphorylation of PFKP and PKM2 by cyclin D3–CDK6 kinase, a core complex in the cell cycle machinery8. The inhibition of these glycolytic enzymes results in an increased production of NADPH and GSH8.
Additionally, an increased glutamine uptake has been observed in many types of cancer4, 43, 44, 45. In the study by Yoo et al. (2020), an increased uptake of glutamine through glutamine-supported OXPHOS supplied the ATP demand of the highly proliferative pancreatic cancer cells43. A high glutamine level also increases the cellular GSH level, which suppresses the production of reactive oxygen species (ROS) and protects cells from chemotherapy damage11, 21, 43. Dysregulated glutamine metabolism is associated with an increased expression of oncogenes, such as c-Myc4. c-Myc induces the overexpression of several genes in the glutamine metabolism pathway, including GLS I and phosphoribosyl pyrophosphate amidotransferase, and consequently mediates the malignant progression of small-cell lung cancer4, 29. A high level of glutamine also increases the cellular GSH level, which minimizes the damage caused by ROS and leads to chemotherapy drug resistance in both cancer cell lines and patient samples11, 21, 43.
Metabolic Rewiring Under Selective Pressure
Many anti-cancer agents target cells in the proliferating stage that are dependent on glycolysis11, 12, 46; however, recent evidence shows that surviving cancer cells escape treatment by shifting their metabolic activities toward other shunt pathways, such as OXPHOS, the PPP, and the SSP11, 12, 14, 47. These cells remain dormant for a certain period and potentially rise and cause relapse in the future12, 14, 47.
Notably, in their studies on acute myeloid leukemia (AML), Jones and colleagues have identified glycolytic-dependent treatment-sensitive and OXPHOS-dependent treatment-resistant leukemia cells11. Interestingly, the OXPHOS-dependent cells did not rely on glucose as the source of nutrients; instead, they were dependent on amino acid metabolism11. Treatment-resistant cells can also dysregulate metabolism toward lipid biosynthesis14, 22. For example, a multi-omics analysis revealed an upregulation of fatty acid biosynthesis accompanied with a downregulation of TCA and amino acid in EZH2 inhibitor-treated cells22. Additionally, invasive breast cancer cells can also shift toward the PPP and SSP when glycolysis is suppressed8, 21.
The mechanism of how cancer cells reprogram their metabolic activity to acquire resistance is thus far poorly understood. Some studies have suggested a compensatory energy acquisition associated with dysregulated expressions of oncogenes in resistant subclones, such as RAS and MAPK12, 14, 44, 48. For example, an RNA-seq analysis on samples from relapsed or resistant AML cases revealed an enrichment of the intrinsic RAS expression in resistant subclones12, 14. RAS can induce mitochondrial fusion that further elevates OXPHOS activity39, 49. Furthermore, when the glycolysis pathway is inhibited, cancer cells direct the glycolytic flux toward the PPP, thereby increasing the NADPH level21. NADPH is essential not only for macromolecule synthesis in the growing phase of the cell but also for protecting cells from oxidative damage. In the presence of NADPH, GSH reductase converts oxidized GSH to reduce the GSH level, which protects cancer cells from oxidative damage caused by ROS. Notably, low levels of ROS have been linked to the stemness properties of resistant cancer cells, explaining the protective mechanism executed by resistant cells11, 21.
Furthermore, the metabolic shift from glycolysis toward lipid metabolism under anti-cancer therapy is linked to cell cycle reprogramming as well as global methylation status modification14, 22, 50. For example, FLT3-mutated AML cells with an altered metabolism escaped the initial chemotherapy-induced G0G1 cell cycle arrest14. In the evolution toward resistancy, sphingolipid or phospholipid metabolism was enriched in surviving cells accompanied by an expansion of NRAS mutation and a more prominent AURKB signaling that drove the cells out from dormancy to become highly proliferative14. Another study crosslinked transcriptomic and metabolomic data of solid tumor samples that have been treated with GSK126, a selective inhibitor of EZH2 methyltransferase22. The analysis revealed that epigenetic modifications were implicated in the treatment-insensitive samples, in which a global reduction of the H3K27me3 level was observed; this thereby increased the expression of target genes in lipid biosynthesis, such as SCD1 and ELOVL222.
TARGETING METABOLISM
The class of drugs resembling the structure of nucleotide metabolites but differing enough to inhibit the metabolic activity is termed antimetabolites. The development of antimetabolites originated from the success of aminopterin used in clinical trials to treat childhood leukemia51, 52. The discovery of aminopterin led to the development of methotrexate and pemetrexed, two folate analogs that inhibit the de novo synthesis of nucleotide53. Methotrexate is used in childhood acute lymphoblastic leukemia chemotherapy treatment, which has been reported to yield a cure rate significantly higher than 80% in a phase III clinical study54. Another synthetic nucleotide used in the treatment of gastrointestinal cancer is 5-fluorouracil (5-FU), an analog of pyrimidine53. 5-FU inhibits the formation of thymidine nucleotides during DNA synthesis. Conversely, purine analogs, 6-mercaptopurine and 6-thioguanine, obstruct the synthesis of purine53. Gemcitabine and cytarabine are nucleoside analogs competitively incorporated into replicating DNA internucleoside, thus inhibiting nucleotide chain elongation; they have been used extensively in treating leukemia55, 56.
Recently, efforts have been made to target the metabolic enzymes or proteins that are differentially expressed in cancer. For example, STF-31 was developed to inhibit GLUT1 activity; however, this compound also inhibits nicotinamide phosphoribosyltransferase, an enzyme involved in NADH biosynthesis57. Another inhibitor of GLUT is Glutor, which targets GLUT1, GLUT2, and GLUT358 . However, Glutor alone is not enough to completely eradicate cancer cells. The combination of Glutor and GLS inhibitor at low concentrations synergistically inhibited HCT116 cell growth, which may offer a promising approach for dual inhibition58. Furthermore, a potent MCT1 inhibitor, AZD3965, has undergone phase 1 clinical study in patients with advanced-stage solid tumors59. The results indicate a promising therapeutic value in patients with a high MCT1 expression, and further clinical trials for this compound are ongoing59. A glutamine transport antagonist was also developed to inhibit the uptake of glutamine through the specific glutamine transporter, ASCT260, 61. The compound known as V-9302 mimics glutamine and is able to hamper cell proliferation and induce apoptosis in vitro in more than half of various cell lines used in the study60. However, an in vivo study showed a non-target effect of V-9302 on other transporters, which impedes further exploration of this compound as a potential therapeutic agent61. A comprehensive review of the drugs used to target metabolism in cancer can be found in this study53.
Nevertheless, specificity remains one of the issues in targeting dysregulated metabolism in cancer. Furthermore, single-drug treatment is usually associated with high toxicity, as high concentrations of drug used during chemotherapy induction further increase the risks of long-term side effects in children and worse clinical outcomes in adult patients12. Single-drug treatment can also cause residual disease, indicating resistance and potential relapse14, 50. Thus, combinations of standard chemotherapy agents with novel compound or drug repurposing approaches may help beneficial therapeutic strategies to overcome these issues, as indicated in many studies10, 11, 12, 14.
CONCLUSION
The global landscape of cancer metabolism has evolved from a simple model of the Warburg effect to more complex models of tumor metabolism. Currently, researchers are exploring metabolic targets and pathways used by cancer cells during tumor evolution, invasion, and resistance that can be exploited as biomarkers for diagnostic purposes. More importantly, the discovery can be manipulated to develop novel inhibitors for targeted therapy that may have the potential to synergize with chemotherapy agents currently used in clinics. Not only will this approach eradicate the metabolic flexible- and resistant-cancer cells but also address the toxicity issue of single-drug treatment in cancer therapy. Consequently, this strategy is hoped to foster a better clinical outcome among patients diagnosed with cancer and further improve the quality of life among survivors.
Abbreviations
ATP: Adenosine triphosphate, CDK6: Cyclin-dependent kinase 6, ETC: Electron transport chain, FADH2: Flavin adenine dinucleotide, GLS: Glutaminase, GLUT: Glucose transporter, GSH: Glutathione, HK: Hexokinase, LDH: Lactate dehydrogenase, MCT: Monocarboxylate transporter, NADH: Nicotinamide adenine dinucleotide, NADPH: Nicotinamide adenine dinucleotide phosphate, NHE-1: Sodium-hydrogen exchanger, OXPHOS: Oxidative phosphorylation, PFK1: Phosphofructokinase 1, PPAT: Phosphoribosyl pyrophosphate amidotransferase, PPP: Pentose phosphate pathway, ROS: Reactive oxygen species, SSP: Serine synthesis, TCA: Tricarboxylic acid
Acknowledgments
None.
Author’s contributions
Not applicable.
Funding
None.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.