

Mature Plasmacytoid Dendritic Cell Proliferation Associated Acute Myeloid Leukaemia: A Case Report and Diagnostic Challenge
- Department of Hematology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
- Hospital Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
- School of Dental Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia
Abstract
Mature plasmacytoid dendritic cell proliferation associated with acute myeloid leukaemia (pDC-AML) is a rare haematologic malignancy marked by clonal proliferation of ≥2% pDCs in bone marrow or peripheral blood. We report a 41-year-old woman with underlying neurofibromatosis who presented with anaemia, persistent fever, and skin lesions. Diagnostic evaluations confirmed pDC-AML based on morphological, immunophenotypic, and genetic findings, including ETV6-PDGFRB fusion. Despite multiple chemotherapy regimens, her disease remained refractory. She ultimately succumbed to febrile neutropenia and pulmonary haemorrhage, with disease progression to over 80% circulating blasts. Differentiating pDC-AML from blastic plasmacytoid dendritic cell neoplasm is crucial due to overlapping morphological features but distinct clinical and immunophenotypic profiles. Accurate diagnosis relies on flow cytometry to highlight the absence of CD56 in pDC-AML. Early recognition and precise characterisation of pDC-AML are essential for optimising management strategies, though prognosis remains poor.
Introduction
Plasmacytoid dendritic cells (pDCs) are haematopoietic cells that are part of the innate immune system, functioning as antigen-presenting cells and recognised for their role as type I interferon-producing cells1, 2. The cells develop from bone marrow (BM) precursors, known as macrophage–dendritic cell progenitors, with differentiation into monocytes, conventional dendritic cells (cDCs) and pDCs1. pDCs constitute less than 0.1% of leucocytes in peripheral blood2.
Proliferation of neoplastic pDCs is very rare and can be classified into two types: blastic plasmacytoid dendritic cell neoplasm (BPDCN) and mature plasmacytoid dendritic cell proliferation (MPDCP). MPDCP is typically associated with myeloid neoplasms, most frequently chronic myelomonocytic leukaemia (CMML), as well as acute myeloid leukaemia (AML) with monocytic differentiation or myelodysplastic syndrome (MDS)3. Morphology assessment, combined with flow cytometry immunophenotyping or immunohistochemistry, is mandatory to differentiate BPDCN and MPDCP. Mature plasmacytoid dendritic cells proliferation‑associated AML (pDC‑AML) can be differentiated from BPDCN by the presence of an immature myeloid population (CD34, CD117, CD123 and lacking pDC markers) alongside an excess of mature pDCs (CD123, CD4, CD56)3.
BPDCN is a clinically aggressive neoplasm with a poor long‑term prognosis, predominantly affecting elderly patients. It is officially referenced in the World Health Organization (WHO) 2017 classification, with MPDCP identified as a differential diagnosis4. MPDCP is then officially referenced in the subsequent WHO 2022 classification5. The significance of pDC‑AML remains poorly understood. A few recent studies indicate that MPDCP‑associated myeloid neoplasms (including pDC‑AML) have adverse outcomes with shorter survival rates and that it serves as an independent prognostic factor for overall survival5, 6.
Given the limited information available on the diagnosis and outcome of pDC‑AML, we present a diagnostically challenging case of pDC‑AML involving a young patient characterised by treatment resistance and poor prognosis. This case highlights the importance of considering BPDCN as a differential diagnosis. We hope this case report will assist haematologists in diagnosing and managing pDC‑AML more effectively in the future, with particular attention to the treatment resistance and poor prognosis associated with the disease.
Case report
A 41-year-old woman presented with symptoms of anaemia (lethargy, and reduced effort tolerance), accompanied by persistent non-productive cough and intermittent low-grade fever for three months. On physical examination, she exhibited pallor, a haematoma on her left cheek and swelling of the upper gums. Abdominal examination revealed mild hepatomegaly with no splenomegaly or palpable lymphadenopathy. Multiple small pedunculated hyperpigmented skin lesions were noted on the back, abdomen, left breast, thigh, and pubic region, suggestive of neurofibromatosis (NF) related skin lesion. She had a significant family history of skin lesions described as café-au-lait spots observed in her younger brother and three of her children.

Peripheral blood film shows circulating blast (red arrow) and monocytes precursors (blue arrow) with abnormal monocytes (green arrow) describe as monocyte with convoluted nuclei, immature appearing chromatin and small nucleoli.
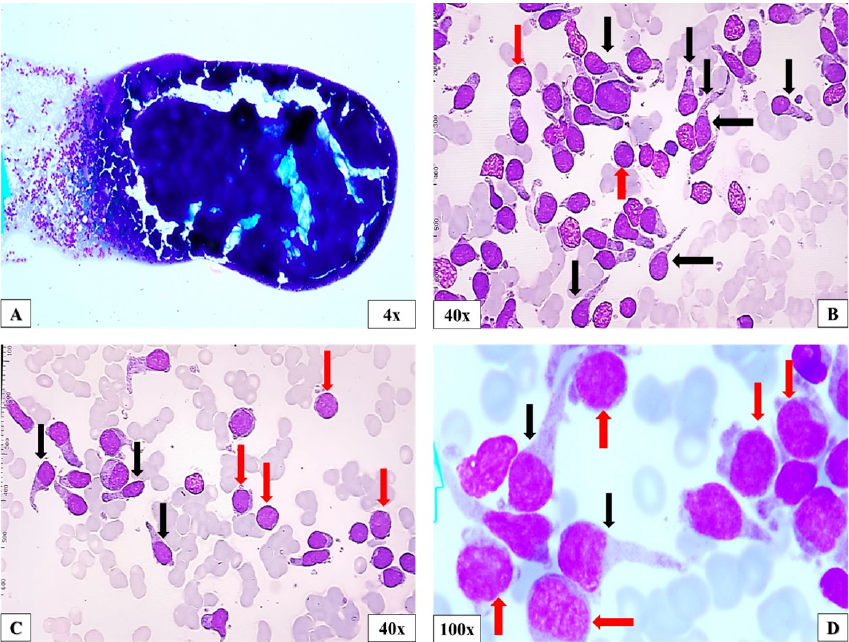
The full blood count revealed severe anaemia (haemoglobin level of 7.0 g/dL), thrombocytopenia (platelet count of 48x10/L) and monocytosis (5.3x10/L). The peripheral blood (PB) film showed mild neutropenia with dysplastic neutrophils (hypogranulation), monocytosis with abnormal monocytes morphology and 15% of circulating blasts (Figure 1). Further laboratory test evaluations, including bone marrow aspirate (BMA) revealed a markedly hypercellular marrow with increased of blasts (myeloblast and monocytic precursors) and proliferation of abnormal mononuclear cells displayed oval clumped nuclei, abundant cytoplasm with long cytoplasmic projections, and increased haemophagocytic activity. Normal haematopoietic cells were markedly reduced (Figure 2) Flow cytometry immunophenotyping identified the myeloblast population (CD34, myeloperoxidase [MPO], CD13, CD117, HLA-DR, CD123, CD64, CD14, CD56) and monocytic precursors (MPO, CD13, CD117, CD33, HLA-DR, CD64, CD14, CD4, CD123, CD34, CD56). In addition, another population that suggest mature pDCs is identified as CD123, CD4, HLA-DR, CD36, CD34, CD117, CD56, MPO, CD64, CD14 (Figure 3).

Bone marrow aspirate.A. markedly hypercellular marrow; B-D. an increase of blast cells (myeloblast and monocytes precursor, indicated by the red arrow), along with the proliferation of abnormal mononuclear cells displaying oval shape and elongated cytoplasmic projection (black arrow), which suggestive of mature pDCs. Normal haematopoietic cells are markedly reduced.

Flow cytometry immunophenotyping of BM aspirate identifies three populations of interest.1. myeloblast (blue population): CD45+dim/low SCC, CD34+, MPO-/+, CD13+, CD117+, HLA-DR+, CD123+dim, CD64-, CD14-, CD56-; 2. monocytic precursor (brown population): CD45+moderate/high SCC, MPO+, CD13+, CD117+, CD33+, HLA-DR+, CD64+, CD4+dim, CD123+dim, CD34-, CD56-; 3. mature pDCs (orange population): CD45+bright/low SCC, CD123+bright, CD4+bright, HLA-DR+, CD36+dim, CD34-, CD117-, CD56-, MPO-, CD64-, CD14-
The trephine biopsy (TB) also revealed marked hypercellular marrow with increased of blast cells (positive for MPO, CD117, nTdT, lysozyme, and negative for CD34, CD3, CD19 and CD68). Additionally, another abnormal mononuclear cell was observed, characterized by oval in shapes and elongated cytoplasm (positive for lysozyme and negative for MPO, CD34, CD117, nTdT, CD3, CD19 and CD68) (Figure 4). The other immunohistochemical (IHC) markers to confirm pDCs (CD123, CD56 and CD4) were not accessible. Cytogenetic analysis by conventional karyotyping revealed a normal 46, XX karyotype, with no clonal numerical or structural chromosomal abnormalities. However, molecular testing using Hemavision-28N variant identified ETV6-PDGFRB fusion gene, t(5;12)(q33;p13). Unfortunately, patient refused for the skin biopsy, which is necessary for further evaluation to confirm NF and to rule out leukemic infiltration.

Trephine biopsy.A, B. marked hypercellular marrow with increased of blast cells and abnormal oval cells with elongated cytoplasm; C-E. The blast and abnormal cells are positive (highlighted by a dark brown stain) for MPO, CD117 (scattered) and lysozyme, and negative for CD68. However, proliferation of pDCs cannot be confirmed by the IHC due to unavailability of CD123, CD56, and CD4 antibody markers.
Based on laboratory and immunophenotypic findings, the patient was diagnosed with pDC-AML. She was treated with multiple chemotherapy regime but showed refractory to treatment. Initially, she received induction therapy with acute myeloid leukaemia 3+7 regime (three days of daunorubicin 50mg/m/day and seven days of cytarabine 100mg/m/day). Since remission was not achieved, she subsequently received reinduction chemotherapy with a regimen consist of five days of mitoxantrone (6mg/m/day), etoposide (80mg/m/day) and cytarabine (1g/m/day) (MEC). However, marrow remission was still not achieved. Due to refractory nature of the disease to these two chemotherapy regimens and the patient’s young age, she was treated with extensive regime consist of five days of fludarabine (30mg/m/day) and cytarabine (2g/m/day), granulocyte stimulating factor (300 g/day until neutrophil recovery) and three days of idarubicin (8mg/m/day) (FLAG-Ida). Unfortunately, the patient succumbed to death due to febrile neutropenia and pulmonary haemorrhage following the FLAG-Ida regime, with a PB film showed over 80% of circulating blast. The patient's condition deteriorated rapidly, leaving insufficient time to initiate tyrosine kinase inhibitor (TKI) therapy or proceed with the workup for allogeneic hematopoietic stem cell transplantation (HSCT).
Discussion
pDCs are typically a minor component of the cellular composition in PB, BM, and lymph nodes; however, they can proliferate in certain disease states. This proliferation can be broadly categorized into three types: 1. non-clonal proliferation, as seen in reactive conditions highlighting their role in various inflammatory conditions (such as Kikuchi-Fujimoto lymphadenopathy, sarcoidosis, systemic lupus erythematosus) and microbial immunity (tuberculosis and toxoplasmosis); 2. clonal malignant proliferation, which occurs in BPDCN; and 3. clonal non-malignant proliferation, observed in MPDCP7, 8. This broad spectrum of associations emphasizes the importance of pDCs as key players in immune regulation and disease pathophysiology.
MPDCP associated with myeloid neoplasm is defined as a clonal proliferation of pDCs with low-grade morphology identified in a defined myeloid neoplasm. It is also referred to as pDC-myeloid neoplasm, associated with pDC expansion and is defined in the current WHO classification within the context of histiocytic/dendritic cell neoplasms5. MPDCP primarily affects elderly patients with a median age of 69 to 72 years, marked male predominance and is frequently associated with CMML, AML, and MDS9.
The diagnosis of AML with myelomonocytic differentiation associated with pDC-AML is a rare and diagnostically challenging entity. pDC-AML frequently arises in association with RUNX1 mutation and is defined as AML with ≥ 2% mature pDCs of non-erythroid nucleated cells in BM and/or PB5. Molecular testing using Hemavison-28N revealed no involvement of RUNX1 fusion gene in this reported case. Instead, ETV6-PDGFRB fusion gene resulting from t(5;12)(q31-33;p13) was detected. In contrast to CMML, where pDCs are typically mature, in pDC-AML, pDCs show a spectrum from early immature pDCs (CD34, CD303 ) to mature pDCs. The immunophenotype of mature pDCs includes expression of CD123, TCF4, CD2AP, SPIB, CD303, CD304, and MX15.
In the initial flow cytometry analysis, pDCs may have been overlooked due to the high presence of myeloblasts with monocytic differentiation, which is consistent with the diagnosis of acute myelomonocytic leukemia. However, the identification of a population with bright CD123 expression led to further evaluation, revealing cells with markers characteristic of mature pDCs. Correlation of BMA findings with flow cytometry results in this case strongly suggests pDC-AML, a disease that may occur de novo or as a transformation from CMML10. This report indicates ETV6-PDGFRB fusion with no involvement of RUNX1 fusion gene by Hemavison-28N. However, these findings do not indicate a discrepancy with the expected features of pDC-AML, as the Hemavision-28N panel does not exclude the possibility of a RUNX1 mutation, which would require separate molecular testing for confirmation. The predominance of mature pDCs and the presence of ETV6-PDGFRB fusion gene, which is commonly reported in CMML, raises the possibility of pDC-AML with myelomonocytic differentiation in this patient that may have arisen from the transformation of underlying CMML.
In pDC-AML, evidence of a clonal relationship between the myeloblasts and the proliferating pDCs was demonstrated by shared immunophenotypic features on flow cytometry (Figure 3) and the presence of the ETV6-PDGFRB fusion within the leukemic population, which has been described in association with both myeloid and pDC lineages. These findings, along with the co-occurrence of AML and mature pDC proliferation, supported the interpretation of a shared clonal origin. While the ETV6-PDGFRB fusion is recognized in various myeloid neoplasms, including CMML and atypical CML, its presence in pDC-AML is considered rare. However, genomic aberrations involving 12p/ETV6 have been identified in BPDCN cases, further supporting the notion that ETV6-PDGFRB fusion can be implicated in both myeloid and pDC lineages11.
To date, no published case reports have documented the co-occurrence of pDC-AML with the ETV6-PDGFRB fusion gene in a patient with a diagnosis of NF. A definitive link between NF and either pDC-AML or the ETV6-PDGFRB fusion gene has not been clearly established. Although the patient exhibited a skin lesion suggestive of NF based on physical examination, the diagnosis was not formally confirmed through genetic testing or biopsy in our setting. NF is known to increase the risk of developing certain blood cancers, particularly juvenile myelomonocytic leukemia (JMML) and other myeloid neoplasms linked to RAS pathway mutations12.
Distinguishing BPDCN from pDC-AML is critical for accurate diagnosis and effective management. Unlike BPDCN, which is characterized by aggressive clinical behavior and frequent skin involvement, pDC-AML typically demonstrates distinct clinical and pathological features. While skin lesions are present in up to 90% of BPDCN cases, pDC-AML is less frequently associated with skin involvement (approximately 25% of cases). Skin lesions in pDC-AML are attributable to either pDC or myeloblast infiltration13. Unfortunately, the patient refused the skin biopsy, preventing definitive histological confirmation of whether the skin lesions were related to NF or due to leukemic or blastic pDC infiltration. This limitation may have impacted the diagnostic assessment of this patient. However, findings from PB, BM studies, and flow cytometry supported the diagnosis of pDC-AML. Nevertheless, based on physical examination, the skin lesions appeared more suggestive of NF-related skin lesions. Other clinical features of BPDCN include generalized lymphadenopathy and/or hepatosplenomegaly, which are often prominent but were absent in this case report.
The overlapping morphological features between MPDCP and BPDCN often make definitive differentiation challenging. A thorough evaluation and interpretation of immunophenotyping are essential for distinguishing these entities. Both BPDCN and MPDCP involve clonal proliferation of pDCs, yet they exhibit distinct immunophenotypic profiles and clinical presentations14. BPDCN cells typically express CD4, CD56, CD123, HLA-DR, TCL1, and TCF4, while lacking lineage-specific markers for B cells (e.g., CD19), T cells (surface and cytoplasmic CD3), and myeloid cells (e.g., myeloperoxidase). Notably, CD56 expression is a hallmark of BPDCN and serves as a key distinguishing feature. Conversely, mature pDCs associated with MPDCP are positive for CD4, CD123, CD303, HLA-DR, and TCF4, but lack lineage-specific markers and do not express CD5613. The absence of CD56 expression in MPDCP provides a critical criterion for differentiation from BPDCN.
Flow cytometry and IHC staining are both effective techniques for evaluating cellular immunophenotype. Flow cytometry is widely accepted as a sensitive and reliable method for identifying pDCs, particularly when interpreted alongside clinical and morphological data. In this case report, although TB could not confirm the pDC proliferation due to the absence of IHC markers such as CD123 and CD56, flow cytometry clearly identified mature pDC proliferation (CD123, CD4, HLA-DR, CD36, CD34, CD56, CD117, MPO, CD64, CD14) as illustrated in Figure 3, supporting a diagnosis of MPDCP rather than BPDCN. Despite this limitation, the diagnosis of pDC-AML was favored over BPDCN based on several factors: the predominant involvement of the bone marrow and peripheral blood with myeloid blasts, the lack of overt blastic morphology among the pDCs, and the absence of a characteristic CD56⁺/CD123⁺ blastic pDC population with high proliferation indices on flow cytometry. Table 1 presents the immunophenotypic characteristics of pDC-AML, BPDCN, and CMML.
Comparison of immunophenotypic markers in pDC-AML, BPDCN, and CMML
|
Marker Expression |
pDC-AML |
BPDCN |
CMML | |
|
Myeloid precursors |
pDC | |||
|
CD45 |
++ |
++ |
+/- |
+++ |
|
CD34 |
++ |
- |
- |
- |
|
CD117 |
+/- |
- |
- |
- |
|
TdT |
+ |
- |
- |
-/+ |
|
CD123 |
-/+ |
+++ |
+++ |
+/- |
|
CD56 |
+/- |
+/- |
+++ |
+/- |
|
CD4 |
+/- |
+/- |
++ |
++ |
|
HLA-DR |
+/- |
+++ |
+++ |
+++ |
|
TCL1 |
- |
-/+ |
++ |
- |
|
CD303 (BDCA-2) |
- |
+/- |
+++ |
- |
|
CD43 |
- |
+ |
+ |
- |
|
CD14 |
+ |
- |
- |
+++ |
|
CD11c |
+ |
- |
- |
+++ |
|
MPO |
++ |
- |
- |
++ |
|
CD33 |
+/- |
- |
+/- |
++ |
|
CD13 |
+/- |
- |
- |
+++ |
This case illustrates the aggressive nature of the disease, characterized by poor prognosis and treatment resistance to multiple chemotherapy regimens in a patient with pDC-AML, as reported in a previous study15. Another previous study reports that AML patients with pDC infiltration showed a higher number of marrow blasts and shorter survival times, which is correlated with a poorer prognosis for AML patients6. The patient did not respond to initial induction therapy with the standard AML 3+7 regimen or the subsequent MEC regimen. As a result, the FLAG-Ida protocol was initiated, aiming to induce remission through intensive chemotherapy. This approach aligned with established guidelines for young patients with aggressive or refractory disease. However, despite intensified treatment, the leukemia remained unresponsive, leading the clinical team to consider allogeneic HSCT as the most viable curative option, given the patient's young age and high-risk features.
A key finding in this case was the presence of the ETV6-PDGFRB fusion gene, a potentially targetable genetic alteration in myeloid malignancies, which can be sensitive to targeted therapy with TKIs like imatinib16. Unfortunately, the patient’s condition deteriorated rapidly, and there was insufficient time to initiate TKI therapy or secure a suitable donor for HSCT. This case highlights the urgent need for early molecular testing to identify targetable mutations promptly and facilitate timely referral for HSCT when indicated.
However, no functional studies were performed to directly evaluate resistance mechanisms such as drug-efflux protein expression or downstream signaling activation from PDGFRB, which could explain the disease’s refractory nature. A previous study found that AML with pDC infiltration exhibited moderate sensitivity to the standard AML chemotherapy regimen and low sensitivity to various other chemotherapeutic agents, suggesting this may contribute to drug resistance and disease relapse6. Additionally, no further molecular testing, such as RUNX1 mutation analysis or advanced molecular study (e.g., next-generation sequencing) to identify other mutations associated with disease aggressiveness or treatment resistance, was performed due to resource limitations at our center. We agree that further genomic profiling could offer valuable prognostic and therapeutic insights, and this will be considered for future cases.
The potential impact of pDC proliferation on the immune microenvironment, including secretion of type I interferons and other cytokines, is recognized as a relevant factor in disease biology and potential treatment resistance. Xiao et al. observed that pDC expansion in RUNX1-mutated AML includes secretion of type I interferons and inflammatory cytokines, which may contribute to shaping the immune microenvironment and potentially impact treatment response, although further mechanistic studies are needed15. However, in this case, functional immune profiling and cytokine measurements (e.g., IFN-α levels) were not available in our institution’s diagnostic repertoire and therefore were not assessed. While we were unable to evaluate these aspects directly, emerging evidence suggests that pDC expansion in myeloid neoplasms may contribute to immune dysregulation and leukemic persistence, which could have been a factor in this patient's aggressive clinical course.
Conclusions
The findings in this case emphasize the importance of integrating morphologic assessment with flow cytometry immunophenotyping and immunohistochemistry for accurate diagnosis. The coexistence of an immature myeloid population (MPO, CD34, CD117, CD123, and lacking pDC markers) with mature pDCs (CD123, CD4, CD56) is a hallmark of pDC-AML. Recognizing the characteristic immunophenotypic and clinical features of pDC-AML allows differentiation from BPDCN and other myeloid neoplasms. Given the poor prognosis and treatment resistance associated with pDC-AML, further research is needed to better understand its biology and optimize therapeutic strategies. This case underscores the need for heightened awareness of pDC-AML among haematologists and pathologists to facilitate earlier diagnosis and tailored management approaches.
Abbreviations
pDCs: Plasmacytoid dendritic cells, BM: Bone marrow, MPDCP: Mature plasmacytoid dendritic cell proliferation, CMML: Chronic myelomonocytic leukaemia, AML: acute myeloid leukaemia, MDS: myelodysplastic syndrome, pDC-AML: Mature plasmacytoid dendritic cells proliferation associated AML, WHO: World Health Organization, PB: Peripheral blood, BMA: Bone marrow aspirate
Acknowledgments
None.
Author’s contributions
Conceptualization: Mohd Nazri Hassan, Shafini Mohamed Yusoff; Collection and processing of material: Mohd Nazri Hassan, Nur Ilyia Syazwani Saidin; Text writing: Mohd Nazri Hassan, Wan Suriana Wan Ab Rahman; Editing: Shafini Mohamed Yusoff, Salfarina Iberahim, Zefarina Zulkafli, Final approval of the version to be submitted: Noor Haslina Mohd Noor, Rosnah Bahar, Marini Ramli All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Ethical approval was not required in accordance with our institution’s policies. The institutional review board (IRB) waived the need for ethical approval because the report describes a single patient case with no experimental intervention, and written informed consent for publication was obtained from the patient.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying image. A copy of the written consent is available for review by Editor-in-chief of this journal.
Declaration of generative AI and AI-assisted technologies in the writing process
ChatGPT was utilized to assist with grammar and language refinement during the preparation of this manuscript.
Competing interests
The authors declare that they have no competing interests.