

Pathophysiology and management of hyperviscosity syndrome as the first oncological emergency in Waldenström’s Macroglobulinemia: A case report and literature review
- Department of Hematology and Medical Oncology, Kermanshah University of Medical Sciences, Kermanshah, Iran
- Department of Hematology and Blood Banking, Faculty of Allied Medicine, Iran University of Medical Sciences, Tehran, Iran
- Department of Genetics, Faculty of Allied Medicine, Shahid Sadoughi University of Medical Sciences, Yazd, Iran
Abstract
Background: Waldenstrom macroglobulinemia (WM) is an exceptionally rare B-cell lymphoma, accounting for less than 2% of all non-Hodgkin lymphomas. Its unique features, including anemia, thrombocytopenia, hepatosplenomegaly, lymphadenopathy, and the occurrence of hyperviscosity, make it a significant focus in the medical community. The extreme rarity of this condition underscores its unparalleled importance and the need for further research. Hyperviscosity syndrome (HVS) is a critical oncological emergency in this disease, leading to increased plasma viscosity and reduced blood flow. Case presentation: We present a case of a 56-year-old man with Waldenström's macroglobulinemia who, following induction therapy, developed a rare hyperviscosity syndrome. Through a combination of plasma exchange therapy and a maintenance program involving Ibrutinib and Lenalidomide, we successfully reduced the patient’s serum viscosity. This successful outcome not only demonstrates the potential for effective treatment strategies in such cases but also instills confidence in the effectiveness of the treatment, offering a beacon of hope for similar cases in the future. Conclusion: The importance of choosing a suitable treatment strategy for Waldenström macroglobulinemia patients is highlighted by the fact that HVS is one of the first oncological emergencies that require timely treatment. This underscores the crucial role of medical professionals, particularly hematologists and oncologists, in promptly diagnosing and treating such cases. The urgency and significance of the issue and the need for swift action are emphasized, as HVS is a condition that cannot be delayed in its management.Introduction
Waldenström's macroglobulinemia (WM) is an exceptionally rare and unique B-cell lymphoproliferative disease characterized by an IgM monoclonal gammopathy and lymphoplasmacytic cell invasion of the bone marrow. Its extreme rarity, accounting for less than 2% of all non-Hodgkin lymphomas, underscores the unparalleled importance of this case in the medical community1. The World Health Organization [WHO] categorization systems classify this medical condition as lymphoplasmacytic lymphoma. An IgM monoclonal gammopathy and lymphoplasmacytic cell invasion of the bone marrow characterize it. Tumor infiltration and excess IgM synthesis are solely responsible for the clinical manifestations of WM2.
At diagnosis, 19% to 28% of individuals with Waldenström's macroglobulinemia (WM) are often asymptomatic. Bone marrow infiltration is always present, and approximately 25% of patients show extramedullary hematopoietic tissue infiltration, causing hepatosplenomegaly and lymphadenopathy3. Due to the displacement of platelets by IgM paraprotein, 25% of patients experience bleeding symptoms such as gingival bleeding and epistaxis3. Hyperviscosity syndrome (HVS) occurs in 30% of WM patients. It is characterized by fatigue, dizziness, and other symptoms, including hearing loss and neurological deficits. Renal involvement can cause proteinuria or nephrotic syndrome, with peripheral neuropathy seen in 20-30% of patients4, 5.
In this case report, we present a WM patient with hyperviscosity syndrome who was unresponsive to routine treatments. However, the introduction of plasma exchange therapy and a maintenance therapy regimen yielded promising results, offering hope for similar cases in the future.
CASE REPORT
In May 2023, a 56-year-old man presented to the Kermanshah Hematology and Oncology Clinic with complaints of fatigue of a few weeks’ duration and weakness, the primary symptoms of concern. The patient had no relevant clinical history. General examination did not show evidence of icterus, generalized lymphadenopathy, or organomegaly. Following a physical exam, the physician requested routine laboratory tests. In primary laboratory investigations, he had normocytic normochromic anemia (MCV 85.6 fL, MCH 70.8 pg) with hemoglobin level: 10 g/dL, normal white blood cell, and a decrease in platelet count (WBC: 4.3 × 10⁹/L; Platelet count: 63 × 10⁹/L). The ESR was 80 mm/h. Renal function tests (RFT) were also performed, and the results showed an increase in blood urea nitrogen (BUN) level (BUN 69 mg/dL, Cr 1.1 mg/dL). ANA and Anti-HCV were nonreactive. Also, the RA factor was negative. Computed tomography (CT) of the abdomen and pelvis determined the absence of organomegaly, and magnetic resonance imaging (MRI) skeletal survey showed no osteolytic lesions. Peripheral blood smear on EDTA showed RBC rouleaux formation. The examination of the bone marrow aspiration (BMA) and bone marrow biopsy (BMB) revealed hypercellular marrow (> 80% cellular marrow) with a marked predominance of lymphoid cell population displaying plasmacytic features (Figure 1). Flow cytometry determined CD19, CD20, and CD138 lymphoplasmacytic cells. Serum protein electrophoresis (SPEP) revealed an M band in the beta-globulin region with an elevated total protein of 9.3 g/dL and an M-spike of 4.6 g/dL. Similarly, Immunofixation electrophoresis (IFE) confirmed IgM-kappa monoclonal protein (Figure 2). MYD88 mutation was detected. Based on these results, the patient was diagnosed with WM/lymphoplasmacytic lymphoma (LPL).

Patient bone marrow biopsy and aspiration. (A) Bone marrow biopsy shows elevated cellularity up to 80% (H&E, ×100). (B) Bone marrow aspiration shows hypercellularity with the predominance of lymphoid cell population with plasmacytic features (Wright-Giemsa staining, ×400).

The serum protein electrophoresis (SPEP) and Immunofixation electrophoresis (IFE) test. A. Initial serum profiles indicate a distinct band in the beta-globulin region during serum capillary zone electrophoresis. B. Immunofixation electrophoresis identified the broad beta-globulin band as an IgM-kappa monoclonal protein.
The patient responded positively to the treatment, immediately starting 6 cycles of the systemic chemoimmunotherapeutic regimens, which included Rituximab, a monoclonal antibody that targets CD20, in combination with Bendamustine-Dexamethasone (RBD). This treatment regimen was chosen based on the patient’s age, overall health, and the stage of the disease. After 1 year of initiating rituximab therapy, the decision was made to transition the patient to a maintenance regimen that combined Ibrutinib and Bortezomib, a choice reflecting his good condition and promising outlook. During the treatment, kidney function tests were normal, and the full blood count (FBC) was as follows: WBC: 5 × 10⁹/L, Hb: 11 g/dL, and Plt: 175 × 10⁹/L.
In July 2024, the patient was suddenly referred to the clinic due to emergency symptoms of hyperviscosity syndrome (HVS), which include dizziness, blurred vision, and resting hand tremors. The total serum IgM was quantified at 4,600 mg/dL, with serum viscosity measuring 4.1 centipoises (cP). The patient also displayed signs of sepsis, neutropenia, and a persistent fever. Accordingly, the patient was promptly admitted and started on empirical broad-spectrum antibiotics (meropenem and vancomycin) due to clinical signs of neutropenic sepsis. Concurrently, therapeutic plasma exchange (TPE) involving 1.5 total plasma volumes (PV) was conducted as scheduled over three days. This timely and integrated therapeutic approach resulted in rapid clinical improvement: the patient became afebrile within 72 hours, neutropenia resolved by day 5 with supportive care, and blood cultures remained negative throughout hospitalization. TPE also led to a marked reduction in serum viscosity to 1.98 cP and a decrease in serum IgM to 1,430 mg/dL.
Given the patient’s genetic profile and the considerable toxicity associated with the rituximab-based regimen, maintenance therapy with ibrutinib combined with lenalidomide was initiated. This approach was carried out following the updated NCCN guidelines and under close monitoring for hyperviscosity syndrome, thereby ensuring patient safety and treatment tolerability throughout therapy. The use of transplantation is limited to Waldenström’s macroglobulinemia because most patients are elderly, and this treatment is not practical for all patients.
At the 6-month follow-up, the patient demonstrated sustained clinical and hematologic stability, with serum IgM levels reduced to 1,380 mg/dL and serum viscosity maintained within the normal range (1.46 cP), consistent with a partial response. No recurrence of hyperviscosity symptoms was observed, and routine laboratory parameters remained within normal limits, supporting the durability and tolerability of the maintenance regimen. A visual summary of the patient’s clinical course, including key diagnostic, therapeutic, and follow-up milestones, is provided in Figure 3. The purpose of this timeline is to clarify the chronological progression of events mentioned in our case report.

Clinical timeline of a 56-year-old patient with Waldenström macroglobulinemia. The figure illustrates major clinical events including the initial presentation (May 2023), RBD-based induction chemotherapy, transition to maintenance therapy (May 2024), emergency admission for hyperviscosity syndrome (July 2024), plasma exchange, and sustained clinical response at 6-month follow-up (January 2025).
DISCUSSION
Waldenström macroglobulinemia (WM) is classified as a lymphoplasmacytic lymphoma notable for IgM monoclonal protein. The principal methodology utilized for diagnosing Waldenström macroglobulinemia involves the identification of IgM monoclonal gammopathy alongside findings from bone marrow biopsies6. A remarkable finding is that more than 90% of patients with WM possess the MYD88 L265P mutation, highlighting the significance of this mutation in understanding the disease7. Currently, treatment modalities for WM encompass plasmapheresis, chemotherapy, and various targeted therapeutic interventions8. According to NCCN guidelines, Rituximab-based regimens are the cornerstone of Waldenström macroglobulinemia treatment. However, it is essential to be aware that these therapies may inadvertently worsen hyperviscosity syndrome due to the swift surge in IgM resulting from the destruction of tumor cells. Understanding this balance is crucial for effective management and care9, 10. Mutations in MYD88 are linked to the ongoing activation of the BTK pathway, making BTK inhibitors a valuable treatment option. The B-cell antigen receptor and Bruton’s tyrosine kinase (BTK) pathways are essential for B-cell malignancies like Waldenström macroglobulinemia (WM). Currently, ibrutinib is the sole approved BTK inhibitor for this condition, bringing renewed hope and promises of better outcomes for patients battling Waldenström macroglobulinemia11. The International Prognostic Scoring System for WM (ISSWM) identifies five key unfavorable factors: (1) Age over 65 years, (2) Hemoglobin below 11.5 g/dL, (3) Platelet count <100×10⁹/L, (4) Serum β2 microglobulin exceeding 3 mg/L, (5) Serum monoclonal protein greater than 7 g/dL.
Patients with two or more of these factors have a five-year survival rate of only 36%, while those with zero to one factor have a significantly higher survival rate of 93%12. The management of WM poses considerable challenges, as no singular therapeutic approach has been established as a definitive cure⁸. This underscores the crucial role of medical professionals and researchers in hematology and oncology, who are at the forefront of developing and implementing these treatment modalities and whose work is invaluable to the field.
The management of Waldenström macroglobulinemia (WM) in this case involved a stepwise therapeutic approach starting with a first-line chemoimmunotherapy regimen comprising rituximab and bendamustine, consistent with NCCN 2024 guidelines for symptomatic WM10. Following hyperviscosity syndrome (HVS), the patient received urgent therapeutic plasma exchange (TPE), leading to rapid improvement and reduced serum IgM levels. Due to treatment-related complications, including neutropenic sepsis, maintenance therapy was subsequently transitioned to an oral regimen consisting of ibrutinib and lenalidomide, tailored to the patient's clinical status and molecular profile (MYD88 L265P-positive). The use of ibrutinib in this patient aligns with current recommendations and clinical evidence supporting its efficacy in MYD88 L265P-mutated Waldenström macroglobulinemia13. In the final analysis of the INNOVATE trial, ibrutinib demonstrated high overall response rates (ORR > 90%) and progression-free survival (PFS) rates exceeding 70% at four years in previously treated WM patients with MYD88 mutations11. While lenalidomide is not part of the standard WM treatment algorithms, emerging evidence from the RV-WM-0426 trial demonstrated its clinical activity with an acceptable safety profile in relapsed/refractory WM patients, achieving an overall response rate (ORR) of 29% and a median time to progression of 16 months, with manageable hematologic toxicities despite prior treatment failures14.

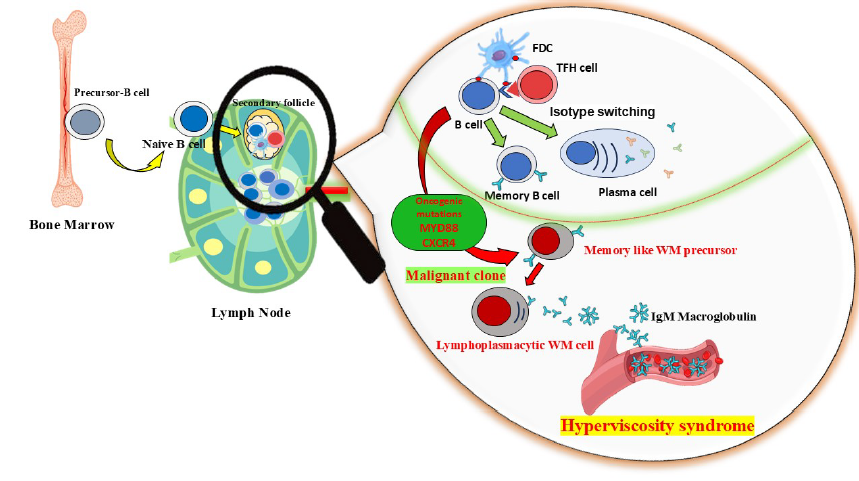
Schematic representation of the pathophysiological cascade in Waldenström macroglobulinemia. Somatic mutations in MYD88 and IgHV lead to clonal B-cell expansion and excessive IgM secretion. The high molecular weight of IgM increases plasma viscosity and promotes red blood cell aggregation, impairing microcirculatory flow and elevating intravascular oncotic pressure. These changes contribute to hallmark clinical signs of hyperviscosity syndrome, including neurologic dysfunction, visual impairment, and mucosal bleeding.
The pathogenesis of Waldenström macroglobulinemia (WM) involves clonal expansion of B lymphocytes harboring somatic mutations in key genes such as MYD88 and IGHV, which arise during isotype switching and somatic hypermutation within lymphoid tissues. These malignant clones produce high levels of pentameric IgM, a large immunoglobulin that interacts electrostatically with erythrocytes, promoting rouleaux formation and increasing plasma oncotic pressure15, 16. As illustrated in Figure 4, this sequence of events reduces microvascular blood flow and contributes to the clinical features of hyperviscosity syndrome, including venous congestion, neurologic deficits, and visual disturbances. The first description of HVS came in 1944 when a published report by Jan Waldenström on two patients with the disease was attributed to him. HVS rarely manifests in patients diagnosed with WM due to the timely identification of the disorder16. This underscores the critical importance of timely diagnosis and treatment of HVS to prevent severe manifestations, highlighting the urgency and importance of your work17. The symptoms associated with hyperviscosity predominantly arise from shear forces that compromise unsupported venous channels, thereby impairing microcirculation. This syndrome is often delineated by a “classic triad” comprising visual impairments, neurological disturbances, and hemorrhagic events4, 18.
Therapeutic plasmapheresis, or plasma exchange, is a medical procedure that separates plasma from blood cells to remove harmful substances19. A key reason for this intervention is managing hyperviscosity syndrome (HVS), often seen in Waldenström macroglobulinemia (WM). The application of plasmapheresis is warranted in symptomatic patients or those experiencing end-organ dysfunction attributable to HVS20. It is crucial to note that plasmapheresis does not modify the fundamental disease process; hence, chemotherapy is typically initiated concurrently. This procedure is notably advocated to precede chemotherapy in patients necessitating prompt paraprotein regulation, akin to the case of our patient. Previous research shows that plasmapheresis can expediently alleviate most clinical manifestations associated with serum HVS, underscoring the critical importance of timely diagnosis and highlighting the urgent nature of our work in managing HVS21.
This manuscript presents a detailed account of a middle-aged male individual diagnosed with Waldenström macroglobulinemia (WM) following the onset of symptoms indicative of fatigue and cytopenia. After one year of therapeutic intervention, the patient experienced the onset of severe hyperviscosity syndrome (HVS), which was substantiated by a markedly elevated serum viscosity level alongside the manifestation of neurological symptoms. The immediate initiation of plasmapheresis effectively mitigated HVS until chemotherapy could be administered to produce a significant therapeutic effect. This case elucidates the varied presentations of WM and highlights the critical importance of the prompt recognition and management of HVS as an urgent oncological emergency.
Additionally, this examination underscores several pivotal considerations about Waldenström macroglobulinemia (WM). First and foremost, a thorough investigation for potential WM should be conducted utilizing diagnostic modalities such as serum protein electrophoresis (SPEP) and bone marrow biopsy, particularly when constitutional symptoms coexist with unexplained cytopenia. This underscores the need for vigilance in our monitoring practices, keeping us alert and attentive. Secondly, it is imperative to vigilantly monitor IgM levels and serum viscosity in patients with an established diagnosis to facilitate the screening for HVS. Thirdly, plasmapheresis is capable of rapidly alleviating symptoms associated with HVS, while chemotherapy is concurrently initiated to secure long-term control of the disease. According to the latest response categories established by the Eleventh International Workshop on Waldenström’s Macroglobulinemia (IWWM-11), the evaluation of therapeutic response hinges on three key factors: the degree of reduction in serum IgM levels, the alleviation of disease-related symptoms, and, in advanced cases, the clearance of both marrow and extramedullary disease. The standardized response categories are as follows: (1) Minor Response (MR): A reduction of 25% to less than 50% in serum IgM levels. (2) Partial Response (PR): A reduction of 50% or more in serum IgM levels. (3) Very Good Partial Response (VGPR): A remarkable reduction of 90% or more in serum IgM levels. (4) Complete Response (CR): Achieving immunofixation negativity in serum, resolution of extramedullary disease, and histological clearance of lymphoplasmacytic cells in the bone marrow22.
In this case, the patient’s serum IgM level impressively decreased from 4,600 mg/dL to 1,380 mg/dL, a striking 70% reduction that clearly fulfills the criteria for partial response (PR). Moreover, the patient experienced a complete resolution of hyperviscosity-related symptoms, which included dizziness, hand tremor, and blurred vision. Notably, serum viscosity normalized from 4.1 cP to 1.46 cP, reinforcing the strength of this response classification. At the 6-month follow-up, no clinical or laboratory evidence indicated disease progression, thereby confirming the durability and efficacy of the therapeutic response. Finally, it is crucial to underscore that WM remains an incurable condition; nevertheless, the paramount objective is to ensure the patient’s quality of life, with prognosis contingent upon risk stratification at the point of diagnosis.
Our patient exhibits several notable features that differentiate him from similar case reports6, 23 as follows: Asymptomatic Waldenström macroglobulinemia, presenting without clinical symptoms.The age of 56 years is relevant for disease progression.Positive response to plasma exchange therapy effectively alleviates HVS symptoms.
CONCLUSION
Waldenström macroglobulinemia management is challenging because the duration of disease control is unpredictable. While plasmapheresis offers rapid symptomatic relief by reducing IgM levels, it is not curative. Long-term disease control requires timely initiation of appropriate maintenance therapy to prevent recurrence and ensure sustained remission.
Abbreviations
BMA: Bone marrow aspiration, BMB: Bone marrow biopsy, BUN: Blood urea nitrogen, CT: Computed tomography, FBC: full blood count, HVS: Hyperviscosity syndrome, IFE: Immunofixation electrophoresis, LPL: lymphoplasmacytic lymphoma, MRI: Magnetic resonance imaging, RFT: Renal function test, SPEP: Serum protein electrophoresis, WHO: World Health Organization, WM: Waldenström macroglobulinemia
Acknowledgments
The authors are grateful to all colleagues at the Clinic of Hematology and Oncology.
Author’s contributions
Mehrdad Payandeh & Afshin Karami: Literature search, Clinical studies, Data acquisition, Data analysis, Guarantor; Noorodin Karami, Azam Yazdanian: Manuscript preparation, Manuscript review, Figure design; Noorodin Karami: Concepts, Design, Definition of intellectual content; Afshin Karami & Yasaman Pouriafar: Manuscript editing, Referencing by EndNote, Manuscript preparation, Literature search. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Data and materials used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The procedures have been reviewed and approved by the ethics committee. All procedures were conducted under the 1964 Declaration of Helsinki and its subsequent amendments or comparable ethical standards. The patient gave written informed consent to publish this case report and any accompanying images.
Consent for publication
Written informed consent was obtained from the patient to publish this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Declaration of generative AI and AI-assisted technologies in the writing process
No generative AI or AI-assisted technologies were used in the writing, editing, or creation of this manuscript. All work was performed by the authors.
Competing interests
The authors declare that they have no competing interests.