

Galactose-1-phosphate modulates mitochondrial function and Toll-like receptor 2 expression in neonatal skin fibroblast cultures
- Department of Pediatrics, College of Medicine, Kuwait University, Jabriya, Kuwait
Abstract
Introduction: Mitochondrial dysfunction and Toll-like receptors (TLRs) are commonly associated with oxidative stress and apoptotic cell death in metabolic disorders; however, their roles in Classical Galactosemia remain largely unexplored. This study examined the activities of key mitochondrial enzymes, as well as the gene expression of peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α) and Toll-like receptor-2, under hypergalactosemic or hyperglycemic conditions in fibroblast cultures.
Methods: Confluent fibroblast cultures were established from the post-circumcision skin of 2–3-day-old healthy neonates. Next, these fibroblast cultures were treated with D-Galactose (D-Gal, 0–20 mM), galactose-1-phosphate (Gal-1-P, 0–5 mM), or “high glucose” (HG, 25 mM). Peripheral blood mononuclear cells (PBMCs) were isolated from galactosemia patients and control subjects. Activities of medium-chain acyl-CoA dehydrogenase (MCAD), cytochrome c oxidase (CcO), and carnitine palmitoyltransferase-1 (CPT-1) were analyzed in cell homogenates and in PBMCs of both patients and controls. ATP production and malondialdehyde (MDA) levels in fibroblasts were measured using commercial kits. In addition, PGC-1α and TLR-2 were examined in fibroblast cultures by western blot analysis.
Results: D-Gal and Gal-1-P significantly (p < 0.05) reduced the enzyme activities of CPT-1, MCAD, and CcO, with Gal-1-P causing a more pronounced (p < 0.01) effect on CPT-1 compared to the control group. HG had no significant impact on these mitochondrial enzymes. Moreover, mitochondrial enzyme activities were markedly lower in PBMCs from galactosemia patients than from control subjects. Galactose-treated fibroblasts showed elevated TLR-2 levels but unchanged PGC-1α expression compared to control cells. Only Gal-1-P significantly diminished PGC-1α levels, whereas HG, D-Gal, and Gal-1-P significantly upregulated the expression of TLR-2.
Conclusion: These findings suggest that galactosemia pathogenesis involves Gal-1-P–mediated mitochondrial dysfunction and elevated TLR-2 gene expression.
Introduction
Cellular metabolic activities, including mitochondrial energy production, regulate the growth and development of tissues and organs in mammals1, 2. The development of oxidative stress and its impact on cell function and survival have been the focus of many studies related to the pathogenic mechanisms of several diseases, such as diabetes, cancer, and cardiovascular disorders3, 4. Mitochondria, the cellular powerhouse, play a significant role in cell function and survival through selective nutrient metabolism and ATP production via oxidative phosphorylation (OXPHOS), oxidation of metabolites through the Krebs cycle, or β-oxidation of fatty acids. Impairment of these key mitochondrial functions has been reported in relation to aging and metabolic disorders, including diabetes5, 6.
Mitochondrial dysfunction is known to be a determining factor for cell survival amid redox imbalance during pathogenesis. Moreover, a significant decline in cellular metabolic activities during diabetes-induced nephropathy has been linked to impaired mitochondrial biogenesis, as evidenced by decreased gene expression of peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC-1α)7. Although previous studies have shown that oxidative stress is associated with galactosemia, it remains to be explored whether the pathogenesis of this inherited metabolic disease involves mitochondrial dysfunction. Galactose is an essential energy source in the early stages of human life, and genetic defects in its metabolic pathways lead to major inherited metabolic diseases8. Classic galactosemia, also known as galactosemia type I, is a severe form of galactosemia that results from a deficiency in Galactose-1-phosphate-uridyltransferase (GALT), leading to the accumulation of galactose and its metabolites, particularly Gal-1-P9.
Earlier studies have reported that Gal-1-P possesses cytotoxic potential and impairs various cellular functions by inducing oxidative stress10, 11. The activities of key antioxidant enzymes, such as catalase, glutathione peroxidase, and superoxide dismutase—responsible for the degradation of various reactive oxygen species (ROS), including hydrogen peroxide and superoxide anions—have been reported to be altered under galactosemic conditions12, 13. It has also been reported that mitochondrial dysfunction induces apoptosis in various cells and tissues under different pathological conditions14.
Toll-like receptors (TLRs), initially characterized as pattern recognition receptors for the innate immune response, are now known to interact with various endogenous proteins and impact intracellular signaling processes involving autophagy and apoptosis15. TLRs are expressed by a variety of cell types, ranging from macrophages to vascular cells and skin fibroblasts. Several reports provide evidence for a connection between TLR signaling and autophagy, suggesting that selective autophagy may be induced as a protective mechanism against free fatty acid-induced cell dysfunction in type 2 diabetes16. Increased TLR-2/TLR-4 expression, signaling, ligands, and functional activation have been reported in diabetic subjects. Currently, there are no reports in the literature establishing any role of mitochondrial dysfunction or TLR expression in relation to galactosemia. This study was conducted to examine the comparative effects of hyperglycemic and hypergalactosemic conditions on mitochondrial metabolism and TLR-2 gene expression.
Methods
Materials
Bovine serum albumin (BSA), penicillin/streptomycin, and fetal bovine serum (FBS) were procured from Sigma Chemical Company (St. Louis, MO). DMEM-Ham’s F-12 (1:1) and trypsin-EDTA were obtained from GIBCO (Grand Island, NY). Primaria tissue culture plates were acquired from Falcon Becton Dickinson (Oxnard, CA). All other reagents were of the highest available quality and purchased from Sigma or Calbiochem.
Methods
This study was approved by the Health Sciences Center Ethical Committee for the use of human subjects in research. Galactosemia patients with a confirmed deficiency of Galactose-1-phosphate uridyltransferase (GALT) in their erythrocytes, along with age- and sex-matched controls, were recruited after obtaining informed consent. For the isolation of PBMCs, blood samples were collected from patients and controls under ethical approval from the Institutional Ethical Review Committee of Kuwait University. PBMCs were isolated using Ficoll gradient centrifugation as previously described17. Foreskin samples from healthy neonates (2-3 days old) were collected to establish fibroblast cell cultures for in vitro studies. Fibroblast cultures were maintained in DMEM-F12 medium supplemented with 10% FBS and penicillin/streptomycin (5 U and 5 µg/ml, respectively) in a humidified atmosphere of 5% CO–95% air. All cultures were assayed for GALT enzyme activity to exclude classical galactosemia before carrying out any experiments.
Treatment of cell cultures with experimental agents
Confluent cell cultures were washed twice with RPMI 1640 medium before initiating each experiment. Cells were treated with high glucose (25 mM) and supraphysiological concentrations of D-galactose (10 mM) or Gal-1-P (5.0 mM) for 24-72 hours, with medium changes every 24 hours. Lipofectamine, a permeation agent that delivers the polar molecule Gal-1-P, was added to both experimental and control groups in certain experiments. Mannitol (0-20 mM) was included in parallel control experiments to assess the effect of osmotic changes on the parameters of interest, as previously reported18. After treatment, cells were harvested and homogenized in 50 mM Tris-HCl buffer (pH 7.4) containing protease inhibitors.
Enzyme assays
Enzyme activities of the key mitochondrial enzymes MCAD, CPT, and CcO were determined in cell homogenates as previously described19, 20, 21. To assay CcO, a known aliquot of cell homogenate was added to an isosmotic medium containing 10 mM KHPO (pH 6.5), 50 mM KCl, 0.25 M sucrose, 1 mg/ml BSA, and 2.5 mM n-dodecylmaltoside, and the reaction was initiated by adding 0.2 mM ferrocytochrome c. CcO activity was evaluated spectrophotometrically at 550 nm by measuring the oxidation of ferrocytochrome c to ferricytochrome c. MCAD activity was examined using a spectrophotometric assay that measures the formation of cinnamoyl-CoA from 3-phenylpropionyl-CoA in the presence of phenazine methosulfate. CPT activity was likewise measured spectrophotometrically by adding a known amount of homogenate to a reaction mixture containing 50 mM Tris-HCl (pH 8.0), 1.25 mM EDTA, 0.1% Triton X-100, 0.25 mM dithiobis-2-nitrobenzoic acid (DTNB), 37.5 mM palmitoyl-CoA, and 1.25 mM L-carnitine, and monitoring CoA-SH release from palmitoyl-CoA at 412 nm.
Measurement of ATP levels and lipid peroxidation (MDA content)
To measure ATP and MDA levels, confluent treated and untreated cells were collected and homogenized in Tris-HCl buffer. ATP content was quantified using a luminescence-based assay kit (ThermoFisher Scientific). The malondialdehyde (MDA) levels, serving as an indicator of lipid peroxidation, were determined with specific commercial assay kits following the manufacturer's protocols.
Western blot analysis
For Western blot analysis, cells were homogenized in lysis buffer containing a protease inhibitor cocktail, then centrifuged at 12,000 rpm for 15 minutes at 4°C. The resulting supernatants were collected, and protein concentration was determined by Lowry’s method. Equal amounts of protein were mixed with loading buffer and resolved on a 10% SDS-PAGE. Separated proteins were transferred onto a PVDF membrane for detection of PGC-1α and TLR-2. After blocking with Blotto B, membranes were incubated overnight with the primary antibodies, washed with Tris-buffered saline containing 2% Tween 20, and then incubated for 2 hours with an HRP-conjugated secondary antibody. β-actin served as a loading control for protein normalization. Protein bands were visualized using SuperSignal chemiluminescent substrate.
Data Collection and Analysis
Data from different groups were analyzed using SPSS software to assess statistical significance. Results are expressed as mean ± S.D. of six independent experiments, and means were compared by t-tests and ANOVA, followed by Bonferroni post hoc tests. A difference was considered significant if the p value was less than 0.05.
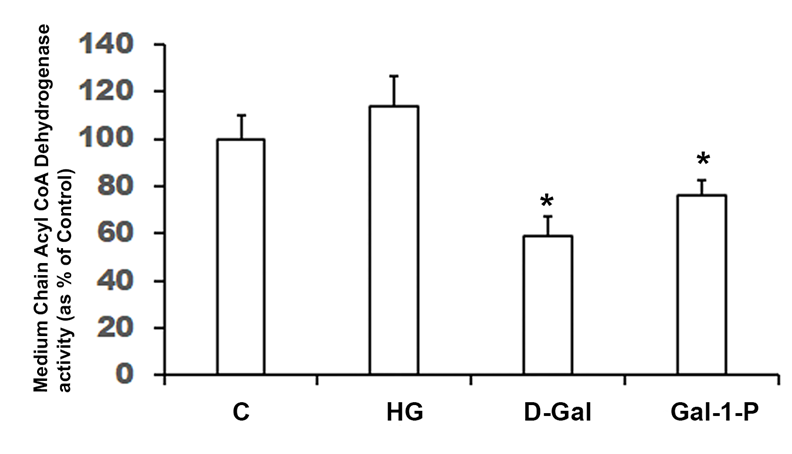
Enzyme activity (shown as percent of control) of Medium-Chain Acyl-CoA dehydrogenase (MCAD) in cell homogenates of skin fibroblasts treated with 25 mM glucose (HG), 10 mM D-Galactose (D-Gal) or 5mM Galactose-1-phosphate (Gal-1-P). Data shown are mean ± standard deviation of six measurements of enzyme assay carried out in duplicate. * p < 0.01 when compared with control (C). Specific activity of MCAD in control fibroblasts was 485.3± 64.4 pmol/min/g protein.
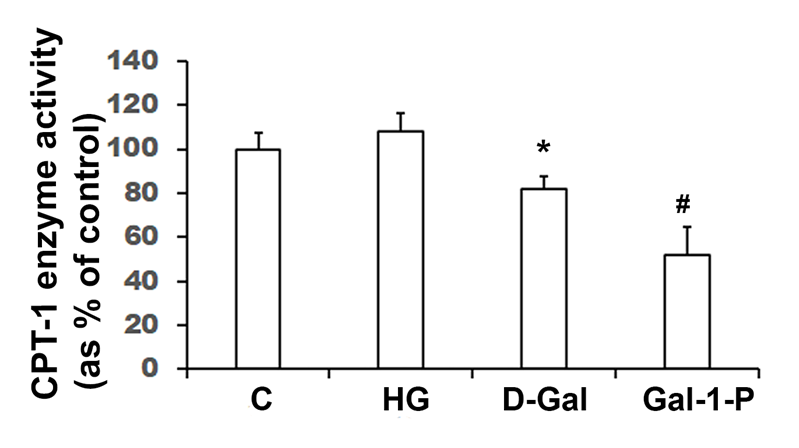
Carnitine Palmitoyltransferase-1 (CPT-1) enzyme activity (shown as percent of control) in homogenates of skin fibroblasts treated with 25 mM glucose (HG), 10 mM D-Galactose (D-Gal) or 5mM Galactose-1-phosphate (Gal-1-P). Data shown are mean ± standard deviation of six measurements of enzyme assay carried out in duplicate. *p < 0.05 and #p < 0.01 when compared with control (C). Specific activity of CPT-1 in control fibroblasts was 38.9 ± 4.1pmol/min/mg protein.
Mitochondrial enzyme activities in homogenates of peripheral blood mononuclear cells isolated from Galactosemia patients and normal healthy subjects
|
Enzyme |
Control (n = 6) |
Galactosemia (n = 6) |
|---|---|---|
|
Medium Chain acyl-CoA Dehydrogenase (MCAD) |
185.3 ± 24.4 |
109 ± 16.4* |
|
Cytochrome C oxidase |
2.19 ± 0.18 |
1.32 ± 0.14* |
|
Carnitine palmitoyl CoA Transferase-1 (CPT-1) |
18.9 ± 2.1 |
11.4 ± 1.3* |
Results
Effects of HG, D-Gal , or Gal-1-P on Mitochondrial Function
Enzyme activities of key mitochondrial enzymes were significantly (p < 0.01) reduced in PBMC obtained from galactosemia patients compared to control subjects (Table 1). Treatment of fibroblast cultures with HG did not significantly affect key fatty acid β-oxidation enzymes, MCAD (Figure 1) and CPT-1 (Figure 2), nor the enzyme activity of CcO (Figure 3). However, the specific activities of all mitochondrial enzymes evaluated in this study were significantly reduced by D-Gal (p < 0.01 for MCAD, p = 0.038 for CPT-1 and p = 0.042 for CcO), as well as by Gal-1-P (p = 0.046 for MCAD and p < 0.01 for CPT-1 and CcO). Interestingly, the effect of Gal-1-P on the activities of CPT and CcO was more pronounced than in control and HG/D-Gal-treated cells. As shown in Figure 4, treatment of fibroblast cultures with HG, D-Gal, and Gal-1-P caused a marked increase in lipid peroxidation, represented by significantly elevated (p < 0.05) MDA levels. However, ATP production was significantly (p < 0.5) impaired only by Gal-1-P, as ATP levels remained unaffected in control cells or those treated with HG or D-Gal (Figure 4).
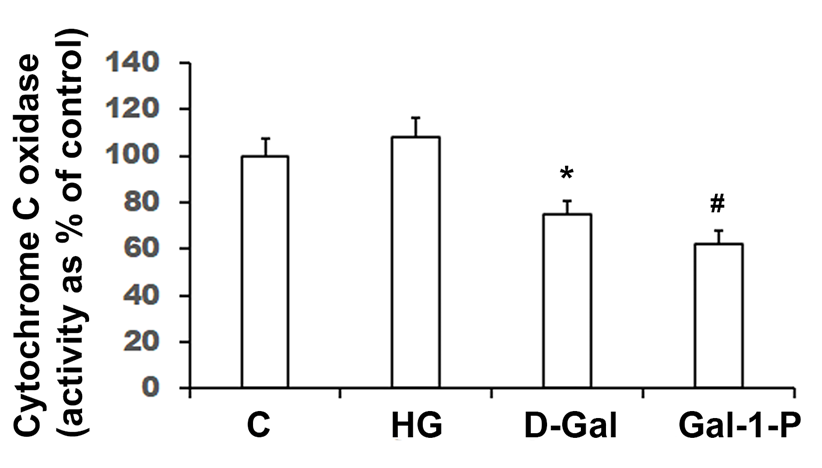
Cytochrome c oxidase (CcO) activity (shown as percent of control) in homogenates of skin fibroblasts treated with 25 mM glucose (HG), 10 mM D-Galactose (D-Gal) or 5mM Galactose-1-phosphate (Gal-1-P). Data shown are mean ± standard deviation of six measurements of enzyme assay carried out in duplicate. *p < 0.01 and #p < 0.01 when compared with control (C). Specific activity of CcO in control fibroblasts was 4.160 ± 0.317 nmol/min/mg protein.
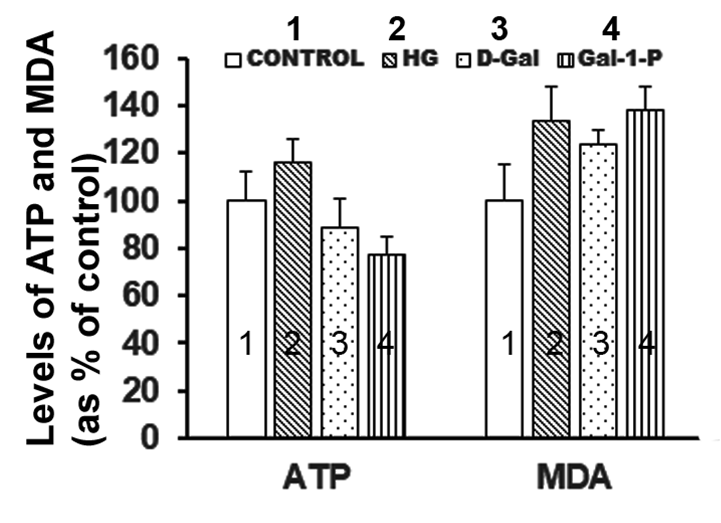
Levels of ATP and MDA (shown as percent of control) in skin fibroblasts treated with 25 mM glucose (2, HG), 10 mM D-Galactose (3, D-Gal) or 5mM Galactose-1-phosphate (4, Gal-1-P). Data shown are mean ± standard deviation of six measurements carried out in duplicate. *p < 0.01 when compared with control (1, C).
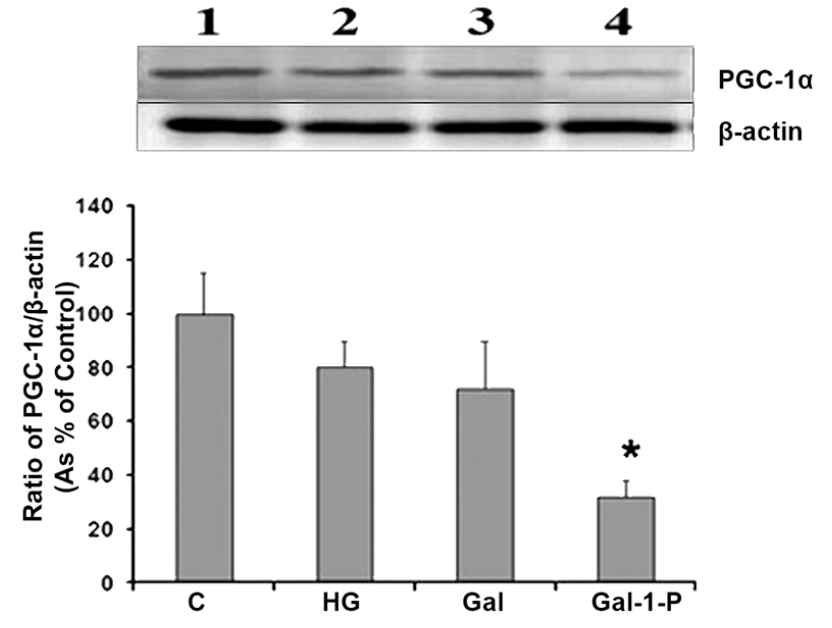
PGC1α expression in cells treated with HG, D-Gal or Gal-1-P
Figure 5 shows the effect of HG, D-Gal, and Gal-1-P on protein levels of PGC1α, a key regulator of mitochondrial biogenesis and hence cellular metabolic state. Both HG and D-Gal did not have any significant effect on the expression of PGC1α, whereas Gal-1-P significantly (p < 0.01) reduced its protein levels, establishing that D-Gal-induced impairment of mitochondrial function is likely mediated through Gal-1-P.
Effect of HG, D-Gal or Gal-1-P on TLR-2 expression
HG, D-Gal, and Gal-1-P were observed to significantly increase (p < 0.01) TLR-2 protein content compared with control cells (Figure 6).
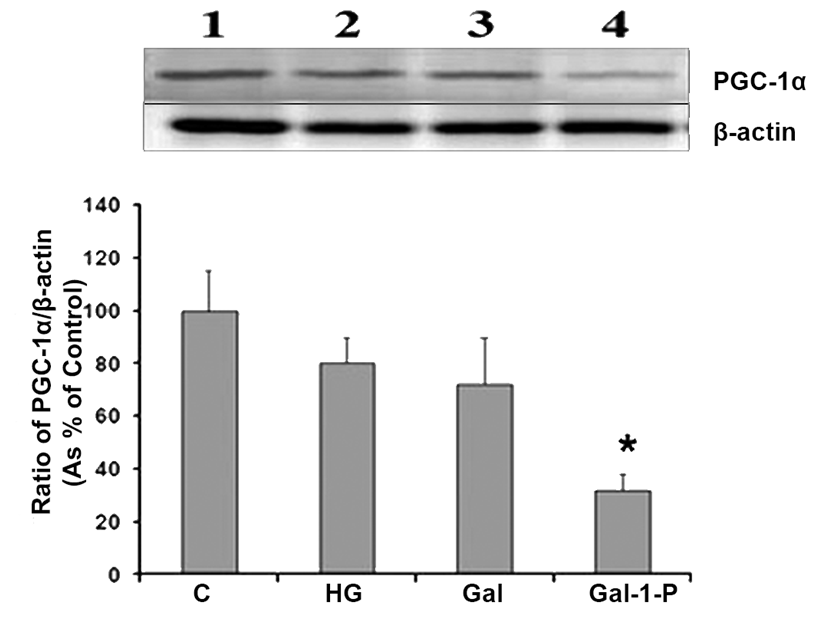
Western blot analysis for detection of PGC-1α levels in skin fibroblasts treated with 25 mM glucose (HG), 10 mM D-Galactose (D-Gal) or 5mM Galactose-1-phosphate (Gal-1-P). β-actin was used as loading control for normalization. Data shown are mean ± standard deviation of the ratio of the protein levels of PGC-1α and β-actin from six experiments. *p < 0.01 when compared with control (C).
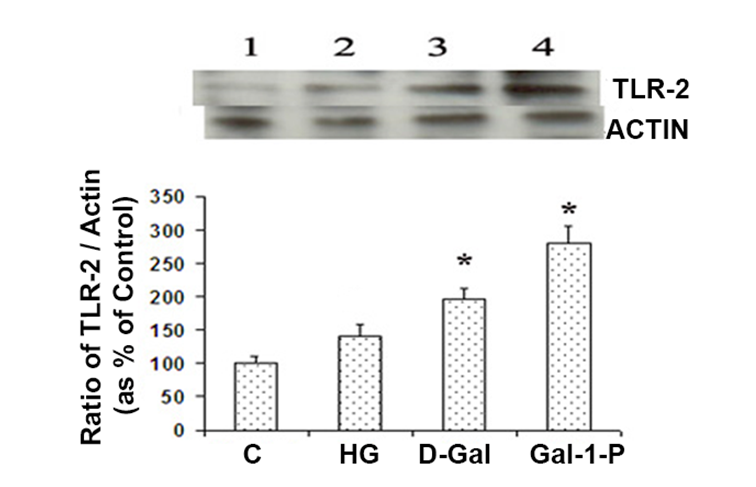
Toll-like receptor-2 (TLR-2) levels in skin fibroblasts treated with 25 mM glucose (HG), 10 mM D-Galactose (D-Gal) or 5mM Galactose-1-phosphate (Gal-1-P). Data are presented as the ratio of TLR-2 and β-actin, an internal control. Data shown are mean ± standard deviation of the ratio of the protein levels of TLR-2 and β-actin from six experiments. *p < 0.01 when compared with control (C).
Discussion
This study demonstrates that D-Gal and Gal-1-P significantly impair some key mitochondrial functions and also activate TLR-2 gene expression whereas HG does not notably affect some key enzymes involved in mitochondrial fatty acid oxidation. Mitochondria are vital subcellular organelles that support cell survival by generating energy. Mitochondrial dysfunction and oxidative stress are reported to be major contributors to a broad range of diseases, including cancer, neurodegenerative disorders, metabolic syndrome, obesity, and diabetes22, 23, 24. However, the role of mitochondria in the pathogenesis of galactosemia has remained largely unexplored. Our findings that PBMCs from galactosemia patients show impaired activities of key mitochondrial enzymes strongly indicate that mitochondrial dysfunction may be a contributing factor to the pathogenesis of classical galactosemia. Cell culture experiments further confirmed that D-Gal and Gal-1-P significantly reduce the enzyme activities of key mitochondrial β-oxidation enzymes, specifically MCAD and CPT1, indicating that supraphysiological levels of D-Gal lead to mitochondrial dysfunction and limit energy production derived from fatty acid metabolism. In contrast, HG did not produce a marked effect on mitochondrial β-oxidation enzymes, suggesting that D-Gal and Gal-1-P exert specific molecular effects on mitochondrial proteins. Previous studies have shown diverse effects of hyperglycemia and diabetes on mitochondrial enzymes in various tissues, under experimental conditions where ATP production and overall mitochondrial capacity were enhanced, leading to increased cellular oxidative stress. The unchanged mitochondrial enzyme activities observed after in vitro treatment of fibroblast cultures with HG may stem from tissue-specific responses or limited exposure to HG. D-Gal and Gal-1-P substantially reduced the enzyme activity of CcO, a key component of the mitochondrial respiratory chain. As Gal-1-P induced a more pronounced reduction in MCAD, CPT-1, and CcO activities compared to D-Gal, and also resulted in reduced ATP production, these findings suggest that the mitochondrial dysfunction observed under galactosemia is primarily driven by Gal-1-P. To determine whether the observed mitochondrial dysfunction results from an inhibitory effect of D-Gal or Gal-1-P on biosynthetic machinery, we examined the protein expression of PGC1α, a key regulatory factor in mitochondrial biogenesis. A significant reduction in PGC1α protein content among Gal-1-P-treated cells strongly supports the notion that mitochondrial biogenesis is compromised under galactosemic conditions. By contrast, HG or D-Gal showed no marked effect on PGC1α gene expression, suggesting that the observed mitochondrial impairment is unlikely to result from direct galactosylation or glycosylation of mitochondrial proteins, but rather from a Gal-1-P-triggered signaling cascade. Numerous studies have demonstrated that PGC1α regulates mitochondrial biology across various cells and tissues, as its increased expression raises mtDNA levels, boosts mitochondrial number, and stimulates the gene expression of multiple mitochondrial enzymes25, 26.
This study demonstrates for the first time, that Gal-1-P induces both mitochondrial dysfunction and the downregulation of PGC-1α, thereby providing new insights into the molecular mechanism of galactosemia. Cell survival and death under various pathophysiological conditions are determined by the state of mitochondrial function, and dysregulation of mitochondrial activities such as β-oxidation and respiratory chain reactions might lead to cell death. Mitochondrial dysfunction has been widely reported to induce cell apoptosis through the activation of intracellular signaling, including certain pattern recognition receptors27. TLRs are membrane-bound pattern recognition receptors that can recognize various molecules produced by damaged cells, in addition to binding molecular motifs of pathogens. Pattern recognition receptors for nucleic acids have been reported to cause mitochondrial apoptosis, and TLR-2 is a transmembrane receptor that recognizes a wide range of exogenous and endogenous ligands including lipids, lipoproteins, nucleic acids and heat shock proteins28, 29. Our findings show that the expression of TLR-2 is markedly increased in cells treated with HG, D-Gal or Gal-1-P, suggesting that these metabolites under hypergalactosemic or hyperglycemic conditions might trigger mitochondrial apoptosis through activation of TLRs. HG, D-Gal and Gal-1-P markedly increased the protein levels of TLR-2; however, TLR-2 levels were significantly higher only in Gal-1-P-treated cells compared with the HG- or D-Gal-treated cells. It has been reported that high glucose induces TLR-2 gene expression30, and this study is the first to demonstrate TLR-2 activation by Gal-P, suggesting galactosemia-related pathogenesis may involve pattern recognition receptors. It has also been reported that elevated cellular oxidative stress activates the TLR-2/4 pathway during hyperglycemia31. Galactosemia has also been reported to be associated with increased production of ROS32, and in line with our findings that HG, D-Gal and Gal-1-P markedly increased lipid peroxidation (MDA levels) in fibroblast cultures, the observed significant increase in TLR-2 protein expression, though more profound in response to Gal-1-P treatment, may be linked to elevated ROS levels. It is an intriguing question whether mitochondrial dysfunction in Gal-1-P-treated cell cultures is caused by increased cellular oxidative stress and activation of TLR-2 or vice versa. Excessive production of ROS has been implicated in mitochondrial dysfunction due to oxidative damage to mitochondria as well as to cellular components, such as proteins, lipids, and DNA33. Although future studies may address the limitations of this study to establish a direct link between mitochondrial dysfunction and TLR-2 activation, it is possible that TLR-2 activation by reactive oxygen species has a direct or indirect inhibitory effect on mitochondrial function, as reported previously29, 30.
Conclusions
In conclusion, the significant inhibitory effect of Gal-1-P on the activities of mitochondrial fatty acid β-oxidation enzymes and cytochrome c oxidase, combined with the downregulation of PGC1α, strongly indicates that mitochondrial dysfunction may be involved in the pathogenesis of galactosemia. Our findings further establish that Gal-1-P also triggers a remarkable activation of TLR-2, suggesting a possible molecular link between TLR activation and mitochondrial dysfunction under galactosemic conditions. The observed deleterious effects of Gal-1-P on mitochondrial function further underscore the previously reported notion that it is the main D-Gal metabolite responsible for Galactosemia-related pathogenesis, highlighting the need to reduce its accumulation to minimize the development and/or progression of the disease.
Abbreviations
ANOVA – Analysis of variance; ATP – Adenosine triphosphate; BSA – Bovine serum albumin; C – Control (group); CcO – Cytochrome c oxidase; CoA-SH – Coenzyme A thiol form; CPT-1 – Carnitine palmitoyltransferase-1; D-Gal – D-Galactose; DMEM – Dulbecco's Modified Eagle Medium; DTNB – Dithiobis-2-nitrobenzoic acid; EDTA – Ethylenediaminetetraacetic acid; FBS – Fetal bovine serum; Ficoll – Polymer for density gradient centrifugation; Gal-1-P – Galactose-1-phosphate; GALT – Galactose-1-phosphate uridyltransferase; HG – High glucose; HRP – Horseradish peroxidase; KCl – Potassium chloride; KH2PO4 – Potassium dihydrogen phosphate; MCAD – Medium-chain acyl-CoA dehydrogenase; MDA – Malondialdehyde; n – Number of samples (e.g., n=6); OXPHOS – Oxidative phosphorylation; PBMCs – Peripheral blood mononuclear cells; PGC-1α – Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; PVDF – Polyvinylidene fluoride; RPMI – Roswell Park Memorial Institute (cell culture medium); ROS – Reactive oxygen species; SDS-PAGE – Sodium dodecyl sulfate–polyacrylamide gel electrophoresis; SPSS – Statistical Package for the Social Sciences; TLRs – Toll-like receptors; Tris-HCl – Tris(hydroxymethyl)aminomethane hydrochloride; Tween 20 – Detergent used in biochemical assays.
Acknowledgments
Authors would like to thank Ms. Heba Dalvi, Ms. Aya and Mrs. Anita for their technical support.
Author’s contributions
GD, WA and HA: Plan the study, supervise laboratory work, collection and analysis of data, manuscript preparation. MH: Perform circumcision and collect foreskin, data analysis and manuscript editing. All authors read and approved the final manuscript.
Funding
This study was supported by Research Grant (MK 01/16) funded by Research Sector, Kuwait University.
Availability of data and materials
Data sets collected and analyzed during this study are presented in this manuscript and are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the Health Sciences Center Ethical Committee for the use of Human subjects in Research.
Consent for publication
Not applicable.
Declaration of generative AI and AI-assisted technologies in the writing process
The authors declare that they have not used generative AI (a type of artificial intelligence technology that can produce various types of content including text, imagery, audio and synthetic data. Examples include ChatGPT, NovelAI, Jasper AI, Rytr AI, DALL-E, etc) and AI-assisted technologies in the writing process before submission.
Competing interests
The authors declare that they have no competing interests.